| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website https://jocmr.elmerjournals.com |
Review
Volume 16, Number 9, August 2024, pages 411-422

Potential Use of MicroRNA Technology in Thalassemia Therapy
Lantip Rujitoa, e ![]() , Tirta Wardanaa
, Tirta Wardanaa ![]() , Wahyu
Siswandarib
, Wahyu
Siswandarib ![]() , Ita Margaretha
Nainggolanc
, Ita Margaretha
Nainggolanc ![]() , Teguh Haryo
Sasongkod
, Teguh Haryo
Sasongkod ![]()
aDepartment of Genetics and Molecular Medicine, Faculty of Medicine, Universitas
Jenderal Soedirman, Purwokerto, Indonesia
bDepartment of Clinical Pathology,
Faculty of Medicine, Universitas Jenderal Soedirman, Purwokerto,
Indonesia
cClinical Pathology Department, School of Medicine and Health
Sciences, Atma Jaya Catholic University, Jakarta, Indonesia
dDepartment of
Physiology, School of Medicine, International Medical University, Kualalumpur,
Malaysia
eCorresponding Author: Lantip Rujito, Department of Genetics and
Molecular Medicine, Faculty of Medicine, Universitas Jenderal Soedirman, Purwokerto, Indonesia
Manuscript submitted June 27, 2024, accepted August 17, 2024, published online August 22,
2024
Short title: MiRNA and Thalassemia Therapy
doi:
https://doi.org/10.14740/jocmr5245
| Abstract | ▴Top |
Thalassemia encompasses a group of inherited hemoglobin disorders characterized by reduced or absent production of the α- or β-globin chains, leading to anemia and other complications. Current management relies on lifelong blood transfusions and iron chelation, which is burdensome for patients. This review summarizes the emerging therapeutic potential of modulating microRNAs (miRNAs) to treat thalassemia. MiRNAs are small non-coding RNAs that regulate gene expression through sequence-specific binding to messenger RNAs (mRNAs). While they commonly repress gene expression by binding to the 3' untranslated regions (UTRs) of target mRNAs, miRNAs can also interact with 5'UTRs and gene promoters to activate gene expression. Many miRNAs are now recognized as critical regulators of erythropoiesis and are abnormally expressed in β-thalassemia. Therapeutically restoring levels of deficient miRNAs or inhibiting overexpression through miRNA mimics or inhibitors (antagomir), respectively, has shown preclinical efficacy in ameliorating thalassemic phenotypes. The miR-144/451 cluster is especially compelling for targeted upregulation to reactivate fetal hemoglobin synthesis. Advances in delivery systems are addressing previous challenges in stability and targeting of miRNA-based drugs. While still early, gene therapy studies suggest combinatorial approaches with miRNA modulation may provide synergistic benefits. Several key considerations remain including enhancing delivery, minimizing off-target effects, and demonstrating long-term safety and efficacy. While no miRNA therapies have yet progressed to clinical testing for thalassemia specifically, important lessons are being learned through clinical trials for other diseases and conditions, such as cancer, cardiovascular diseases, and viral. If limitations can be overcome through multi-disciplinary collaboration, miRNAs hold great promise to expand and transform treatment options for thalassemia in the future by precisely targeting pathogenic molecular networks. Ongoing innovations, such as advancements in miRNA delivery systems, improved targeting mechanisms, and enhanced understanding of miRNA biology, continue to drive progress in this emerging field towards realizing the clinical potential of miRNA-based medicines for thalassemia patients.
Keywords: Thalassemia; Transfusion therapy; MiRNA; Erythropoiesis; MiRNA-based therapy
| Introduction | ▴Top |
Thalassemias are a heterogeneous group of inherited blood disorders resulting from mutations that impair production of the hemoglobin tetramer, which is essential for effective erythropoiesis and oxygen delivery. This is primarily due to reduced or absent synthesis of the α- or β-globin chains that make up hemoglobin. The most severe form, β-thalassemia major, is characterized by severe anemia beginning in infancy, requiring lifelong blood transfusions and extensive iron chelation therapy [1]. Thalassemias are among the most common monogenic diseases worldwide, with over 288,000 annual births affected by severe forms. Globally, carrier frequencies range from 1.5% to > 15% in some regions like the Mediterranean, Middle East, Central Asia, India, and Southern China. Particularly high carrier rates are seen in Cyprus (14%), Sardinia (10.3%), and Southeast [2, 3]. However, rising immigration has increased cases across Europe, North America, and worldwide [4]. Current management of thalassemia major involves chronic blood transfusions usually administered every 2 to 5 weeks along with daily iron chelation therapy to remove excess iron and prevent organ damage. However, these treatments impose considerable burden on patients and are associated with significant treatment-related morbidities [5]. As such, there is an impetus to develop more effective therapeutic approaches for thalassemia that ideally reduce dependence on transfusions and chelators [6]. While thalassemia screening programs are only crucial for prevention, they come with ethical, social, and technical challenges. In regions with high thalassemia prevalence, such as Africa, Southern Europe, the Middle East, and Southeast Asia, the cost and logistical challenges of laboratory diagnostic tests hinder effective screening and diagnosis [7, 8]. Furthermore, the lack of widespread premarital screening and counseling presents a significant obstacle to thalassemia prevention [9].
While blood transfusions and iron chelation remain the standard of care, they are not curative and only ameliorate symptoms without addressing the underlying genetic defects causing thalassemia. Allogeneic hematopoietic stem cell transplantation (HSCT) has long been recognized as the only curative treatment for β-thalassemia, but its application is limited by donor availability and potential complications. More recently, gene therapies have emerged as promising curative approaches. Several novel therapeutic approaches are actively being explored to treat thalassemia more definitively, including gene therapy, genome editing, pharmacological reactivation of fetal hemoglobin (HbF), and modulation of ineffective erythropoiesis [10]. Reblozyl (luspatercept-aamt), a recombinant fusion protein that acts as an erythroid maturation agent, was approved in 2019 by Food and Drug Administration (FDA) for treating anemia in sickle cell patients who require regular red blood cell (RBC) transfusions. This agent represents a significant advance in reducing transfusion burden for some patients [11, 12]. Genome editing through clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) to correct disease-causing mutations in autologous hematopoietic stem cells also holds great potential. The FDA has approved two gene therapies for transfusion-dependent β-thalassemia: betibeglogene autotemcel (beti-cel), a lentiviral vector-based therapy, and exagamglogene autotemcel (exa-cel), a CRISPR/Cas9 gene-editing therapy. These therapies have demonstrated clear efficacy in achieving transfusion independence for many patients, with safety profiles established through clinical trials involving hundreds of patients over last experiments. However, this new approach still needs further optimization, safety evaluation, and validation in large clinical studies [10, 13, 14]. CASGEVY is a groundbreaking gene therapy that has been approved by the FDA as the first CRISPR/Cas9 genome-edited therapy. While the method primarily discusses its application for sickle cell disease (SCD), it is important to note that β-thalassemia is also mentioned [15]. However, it is important to note that access to such novel therapies varies greatly worldwide, with many regions, including parts of Southeast Asia, still relying primarily on traditional management approaches. For instance, in Indonesia, where thalassemia prevalence is high, access to newer therapies remains limited due to cost and infrastructure challenges. This global disparity in treatment availability underscores the ongoing need for developing innovative, accessible therapeutic strategies.
While various innovative genetic and pharmacological approaches are being pursued, one particularly promising and rapidly advancing area is the therapeutic potential of microRNAs (miRNAs) in thalassemia. Several key miRNAs have now been identified that are abnormally expressed in β-thalassemia and drive ineffective erythropoiesis, including miR-15a, miR-16, miR-144, and miR-451 [16, 17]. Therapeutically restoring levels of deficient miRNAs or inhibiting overexpression through miRNA mimics or inhibitors, respectively, has shown preclinical efficacy in reversing thalassemic phenotypes. The miR-144/451 cluster is especially compelling for targeted upregulation to reactivate HbF synthesis. Advancements in delivery systems to increase stability and targeting of miRNA-based drugs are addressing previous challenges. Altogether, modulation of pathogenic miRNAs has emerged as a novel treatment strategy that may offer advantages in specificity, safety, and expansion of therapeutic options for thalassemia patients. Ongoing research and anticipated clinical translation make this an exciting time for developing miRNA therapies [18].
| Development and Biogenesis of MiRNA | ▴Top |
MiRNAs are small non-coding RNAs that play crucial roles in post-transcriptional gene regulation as briefly depicted in Figure 1. The biogenesis of miRNAs in humans involves a series of tightly regulated steps, as depicted. Initially, miRNA genes are transcribed by RNA polymerase II to produce primary miRNAs (pri-miRNAs). These pri-miRNAs contain hairpin structures that are recognized by the Microprocessor complex, consisting of the RNase III enzyme Drosha and the RNA-binding protein DiGeorge syndrome critical region 8 (DGCR8), in the nucleus [19]. Drosha cleaves the pri-miRNAs to release precursor miRNAs (pre-miRNAs). Exportin-5 (XPO5) then transports the pre-miRNAs from the nucleus to the cytoplasm [20]. In the cytoplasm, Dicer processes the pre-miRNAs into mature miRNAs. Various factors influence miRNA biogenesis. For instance, the ubiquitin-specific protease (USP36) has been found to associate with the Microprocessor complex and regulate miRNA biogenesis by SUMOylating DGCR8 [21]. Additionally, specific amino acids in DGCR8 are essential for the interaction with pri-miRNAs and their processing [22].
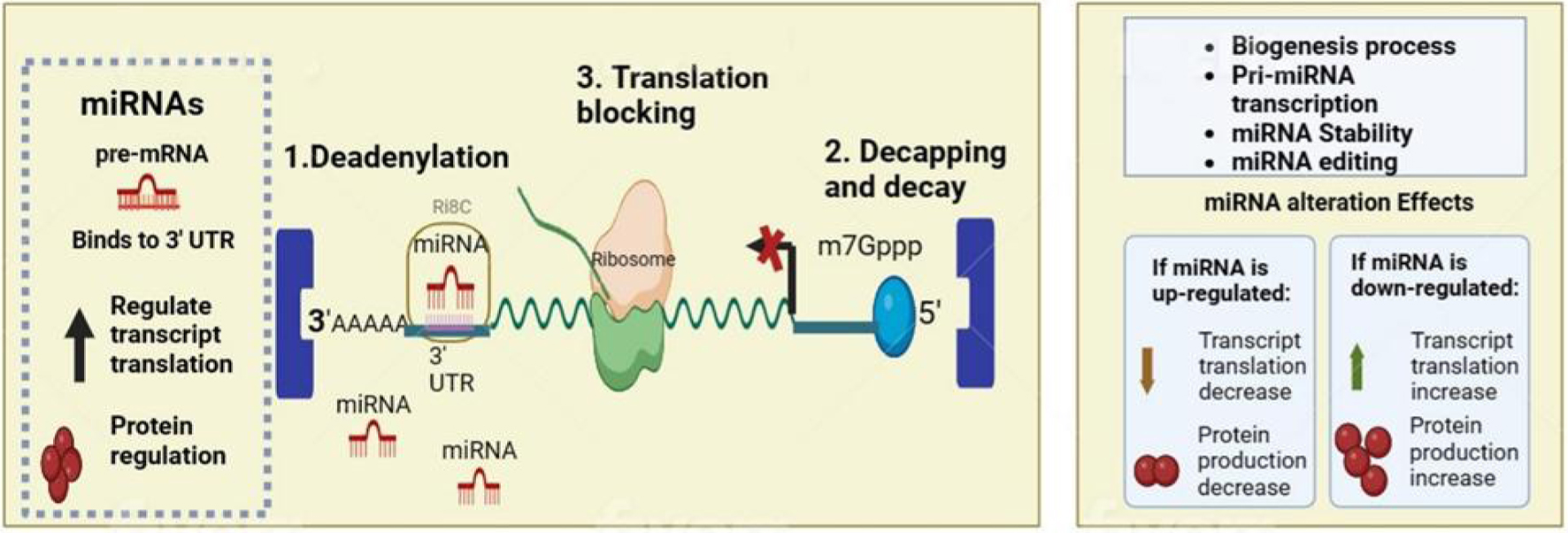 Click for large image |
Figure 1. Normal function and alteration of microRNA (miRNA) in biological mechanism. MiRNAs exert their gene-silencing effects through a sequence of molecular interactions, initiating with deadenylation, where they induce the shortening of the poly(A) tail at the mRNA's 3' end, setting the stage for mRNA degradation; they also facilitate decapping, removing the 5' cap (m7Gppp) which is essential for messenger RNA (mRNA) stability, and they can impede protein synthesis directly by blocking translation, preventing ribosomes from accessing the mRNA. |
| MiRNAs That Are Abnormally Expressed in β-Thalassemia | ▴Top |
Multiple genome-wide profiling studies have identified miRNAs that are differentially expressed in β-thalassemia compared to healthy controls. One of the first miRNAs found to be downregulated is miR-15a, along with other members of its family like miR-16-1, which are markedly decreased in erythroblasts of β-thalassemia patients [23]. Another critical miRNA deficient in β-thalassemia is miR-451, levels of which can influence disease severity based on studies in mouse models. Studies have shown that the loss of miR-144/451 in erythroid cells of Hbbth3/+ mice alleviates β-thalassemia by stimulating unc-51-like kinase 1 (ULK1)-dependent autophagy of free α-globin. This loss of miR-144/451 stimulates ULK1 by activating the liver kinase B1/AMP-activated protein kinase (LKB1/AMPK) axis and inducing erythroblast iron restriction. Therefore, the levels of miR-451 can indeed influence the severity of β-thalassemia [23]. More recent deep sequencing analyses have additionally revealed decreased miR-150, miR-22, and miR-223 as well as upregulated miR-636, miR-576-3p, and miR-642a-5p as signature miRNAs dysregulated in β-thalassemia [23, 24].
With the recognition of their importance in thalassemia pathogenesis, active research continues on profiling miRNA expression changes in β-thalassemia using advanced genomic approaches. Recent next-generation small RNA sequencing analyses have revealed novel miRNAs differentially expressed in β-thalassemia patient erythroid progenitor cells compared to healthy controls. One study uncovered 11 significantly dysregulated miRNAs, including downregulation of miR-486-5p and upregulation of miR-139-5p, miR-30b-5p, and miR-29c-5p [24, 25]. Another sequencing analysis found 14 abnormally expressed miRNAs in β-thalassemia samples, notably decreased miR-618 and increased miR-671-5p. Ongoing work is assessing miRNA profiles in distinct thalassemic cell populations and at different developmental stages [26]. Table 1 illustrates the recap of potential role of miRNA in the pathological aspects of thalassemia.
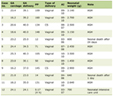 Click to view |
Table 1. List of the MiRNAs Along With Their
Biological Aspects May Be Included in Thalassemia Pathophysiology |
These and other emerging high-throughput studies are uncovering miRNAs previously unknown to be associated with β-thalassemia pathobiology. Capturing the global miRNA expression changes in β-thalassemia will help identify novel regulatory pathways and networks that could be targeted or leveraged for therapy development. Profiling studies are also revealing miRNA signatures that may have prognostic value or predict response to treatments. However, further functional analyses are needed to validate the roles of newly discovered thalassemia-related miRNAs and translate findings from profiling studies into viable therapeutic approaches. Overall, advancing technologies are enabling more comprehensive characterization of miRNA dysregulation in β-thalassemia.
| How Altered MiRNAs Contribute to Ineffective Erythropoiesis | ▴Top |
According to the common definition, ineffective erythropoiesis is defined as the inability to produce a sufficient number of RBCs in the presence of immature erythroid precursors in the bone marrow [27]. This pathological aspect in thalassemia includes increased apoptosis of erythroid precursors, decreased differentiation of erythroid progenitors, increase in oxidative stress, disturbance of iron metabolism, and molecular chaperon dysregulation [28]. Dysregulated miRNAs have been shown to drive ineffective erythropoiesis through targeting and suppressing expression of key proteins involved in erythroid maturation. For example, miR-15a and miR-16-1 downregulate the anti-apoptotic protein Bcl-2, causing increased apoptosis of erythroid precursors [29]. MiR-144 was found to inhibit the erythroid transcription factors GATA-binding factor 1 (GATA1) and T-cell acute lymphocytic leukemia protein 1 (Tal1), leading to blocked maturation [30]. MiR-221/222 were shown to target the kit receptor, impairing erythropoietin signaling. Restoring levels of these deficient or overexpressed miRNAs could potentially correct thalassemic erythropoiesis [31].
More recent findings have uncovered roles of other dysregulated miRNAs in disrupting erythroid maturation in β-thalassemia. MiR-27a was found to be overexpressed and target the erythroid transcription factor KLF8, causing impaired differentiation of erythroblast cells [32]. The miR-99a/let-7e/miR-125a cluster was shown to be upregulated and inhibit multiple parts of the mitogen-activated protein kinase (MAPK) pathway, leading to increased reactive oxygen species and death of thalassemic erythroblasts [32, 33]. Figure 2 illustrates recent findings concerning the role of miRNAs in erythropoiesis. Therapeutic normalization of these and other erythropoiesis-modulating miRNAs could help ameliorate anemia in β-thalassemia. However, better understanding of their myriad downstream targets and complex interactions will be needed to optimize application of miRNA-based drugs.
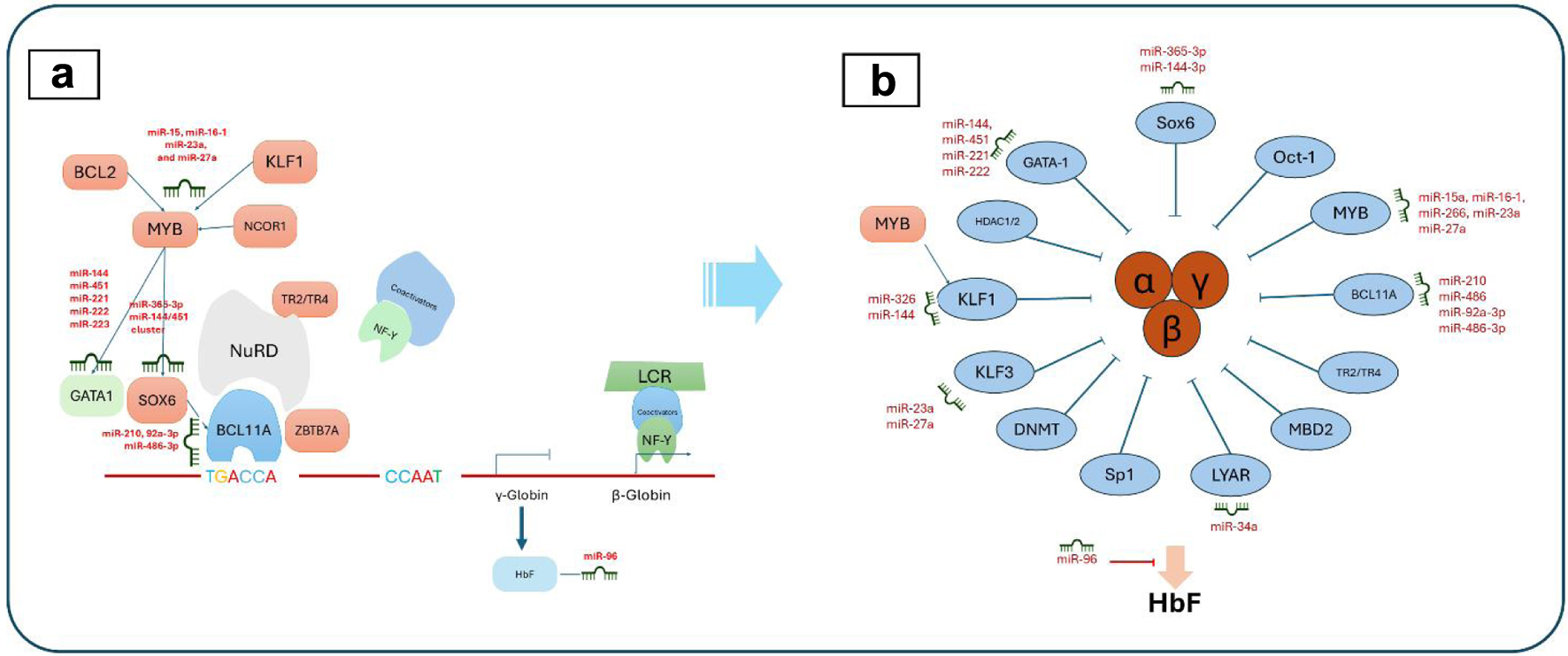 Click for large image |
Figure 2. MicroRNA (miRNA) regulation in erythropoiesis (a) and hemoglobin fetus signaling (b). (a) The complex interplay of transcription factors, miRNAs, and their targets during erythrocyte development, including key regulators such as Myb proto-oncogene protein (MYB), Kruppel-like factor 1 (KLF1), and GATA-binding factor 1 (GATA1). (b) The miRNA-mediated signaling pathways that influence fetal hemoglobin (HbF) expression, highlighting the roles of various factors like KLF1, MYB, and B-cell lymphoma/leukemia 11A (BCL11A) in modulating HbF levels through direct and indirect miRNA interactions. |
| MiRNAs Regulating HbF Expression | ▴Top |
Multiple miRNAs have been implicated in hemoglobin switching and regulation of HbF levels postnatally, as depicted in Figure 2. These include miR-15a/16-1 and miR-150 targeting Myb proto-oncogene protein (MYB), miR-26b-5p regulating Kruppel-like factor 3 (KLF3), and miR-96 affecting γ-globin gene expression. However, the miR-144/miR-451 cluster is considered to have the most therapeutic potential for HbF reactivation, as this locus strongly suppresses HbF during development via B-cell lymphoma/leukemia 11A (BCL11A) and SRY-box transcription factor 6 (SOX6). MiRNA inhibition or overexpression strategies that target this axis may relieve anemia in β-thalassemia through increasing normally silenced HbF [34-37].
Recent functional studies have further clarified mechanisms by which key miRNAs regulate HbF expression. The miR-96 precursor was found to directly target the γ-globin gene promoters, leading to epigenetic silencing mediated by the polycomb repressor complex. MiR-486-3p was shown to suppress HbF by inhibiting erythroid Kruppel-like factor 1 (EKLF) transcriptional activity [38]. The miR-144/miR-451 cluster was found to regulate BCL11A expression via the transcription factors the c-myb proto-oncogene (C-MYB) and SOX6 in later erythroid differentiation stages [30].
Novel genome-wide approaches have also uncovered additional miRNAs that may modulate HbF switching. A functional miRNAs screen identified let-7a-5p as a regulator of BCL11A and HbF expression [39]. Analysis of miRNA and messenger RNA (mRNA) expression changes during erythroid maturation revealed miR-486-3p, miR-92a-3p, and miR-210-5p as candidate regulators of the fetal-to-adult hemoglobin switch. Single-cell miRNA sequencing in differentiating erythroblasts highlighted miR-191-5p as a potential regulator based on its expression patterns [40]. A study identified that hydroxyurea (HU) therapy significantly increases HbF levels in sickle cell anemia (SCA) patients, with miR-210, miR-16-1, and miR-29a acting as positive regulators and miR-96 as a negative regulator of the γ-globin gene. These findings suggest that targeting specific miRNAs could enhance HU-mediated HbF induction, offering potential for improved therapeutic strategies for SCA and thalassemia [41].
These and other emerging findings are elucidating the complex miRNA-mediated networks that orchestrate hemoglobin switching during development. This improved understanding of their mechanisms of action is critical for identifying the most promising miRNA targets to safely reactivate HbF for β-thalassemia therapy. Future studies focused on better defining stage-specific miRNA expression and roles in erythroid subpopulations will further inform therapeutic strategies aimed at HbF induction.
| Current Research on MiRNA-Based Therapies for Thalassemia | ▴Top |
MiRNA mimics to restore normal expression
Therapeutic delivery of miRNA mimics aims to restore normal levels of miRNAs that are deficient or downregulated in β-thalassemia. These synthetic miRNA-like molecules are designed to boost miRNA function by increasing their intracellular concentration. Early studies showed that transfection of mimics for miR-15a and miR-16-1 could reduce ineffective erythropoiesis in cultured thalassemic erythroblasts. A recent report found that mimic replacement of miR-150-5p increased expression of its target MYB, leading to elevated HbF in erythroid progenitor cells from β-thalassemia patients [42].
Other efforts have focused on formulating and delivering miRNA mimics. Packaging miR-15a, miR-16-1, and miR-451 mimics in biodegradable poly-lactic-co-glycolic acid (PLGA) nanoparticles enabled efficient uptake in human thalassemic cell lines and increased miRNA levels for up to 2 weeks post-transfection [43]. Modification with cholesterol has also been tested to improve cellular delivery of miRNA mimics. Cholesterol-conjugated miR-15a and miR-16 mimics demonstrated enhanced erythroid differentiation in vitro and reduced anemia in a thalassemic mouse model. However, challenges remain regarding off-target effects, bioavailability, and developing means to achieve stable miRNA mimic expression [44].
Ongoing research is focused on optimizing miRNA mimics and delivery strategies to restore normal erythropoiesis in thalassemia. Testing combination mimic approaches to target multiple dysregulated miRNAs may offer synergistic benefits. Advancing in vivo testing in animal models will help demonstrate therapeutic potential and inform clinical translation. Overall, mimics present a promising technology but require further innovation to translate into safe and effective miRNA replacement therapies for patients.
Recent advances in nucleic acid chemistry and delivery are enabling further optimization of miRNA mimics. Modified anti-miR-144 mimics containing phosphorothioate backbones exhibited enhanced stability and erythropoiesis-promoting effects in thalassemic mice [45]. Dual-functional miRNA mimics targeting miR-221 and miR-222 with conjugated transferrin improved targeting and uptake into erythroblasts [46]. Mimics encapsulated in platelet membrane-coated nanoparticles showed increased circulation time and delivery to bone marrow after intravenous injection [47]. Testing of self-assembling miRNA mimics is also underway, which may provide advantages in cellular uptake and pharmacokinetics. Overall, chemical and nanotechnology-based strategies are being leveraged to enhance miRNA mimic delivery and activity for therapeutic applications in β-thalassemia.
MiRNA inhibitors/antagomir (antagonis miR) to block abnormal miRNA activity
In contrast to mimics, miRNA inhibitors or antagomir work by blocking the activity of miRNAs that are aberrantly overexpressed in β-thalassemia. These antisense oligonucleotides sequester and silence target miRNAs through complementary base pairing. One of the first antagomir tested was for miR-144, which is upregulated in thalassemic erythroblasts and suppresses GATA1. Transfection of an anti-miR-144 inhibitor improved erythroid maturation and hemoglobin production in cultured patient cells [48, 49].
Recent work has focused on enhancing the stability and delivery of miRNA inhibitors. Modification with locked nucleic acids and 2’-O-methyl groups was found to prolong activity of anti-miR-144 in human thalassemic cell lines [50]. Conjugation with cell-penetrating peptides improved cellular uptake of miR-15a/16-1 inhibitors and reduced their erythroblast toxicity. Anti-miR-221 nanoparticles functionalized with transferrin receptors demonstrated enhanced delivery to thalassemic erythroid precursors. Testing antagomir cocktails targeting multiple dysregulated miRNAs may provide combinatorial benefits [51].
In vivo testing has also shown promise, with short-term antagomir or anti-miR-144 treatment improving anemia in a mouse model of β-thalassemia intermedia [48]. However, challenges remain in achieving stable intracellular activity and prolonged miRNA suppression. Recently, chemically modified anti-miR-144 administered for 3 weeks increased hemoglobin levels and reduced spleen enlargement in thalassemic mice [52]. Further pharmacodynamic analyses and safety evaluations in animal models will be important to support clinical translation.
Overall, miRNA inhibitors represent a promising technology but require additional research to demonstrate durable and specific activity. Advances in chemistry, delivery systems, and testing in vivo will help drive ongoing development of antagomir/antimiRs into clinically viable therapies for rebalancing pathogenic miRNA expression in β-thalassemia.
Using miRNA sponges to inhibit multiple miRNAs in thalassemia therapy
MiRNA sponges are transcripts engineered to contain multiple tandem binding sites for a miRNA or miRNA family, allowing them to stably sequester and inhibit multiple miRNAs simultaneously. Early designs used PCR-amplified sequences with 4 - 10 miRNA binding sites, which were effective in de-repressing miRNA targets. For thalassemia therapy, a sponge targeting miR-15a/16-1 with eight binding sites decreased cytotoxicity and improved maturation in cultured patient erythroblasts [53].
To enhance stability and delivery, miRNA sponges have been incorporated into viral vectors. A lentiviral sponge vector to inhibit miR-221, miR-222, and let-7 significantly increased hemoglobin and erythrocyte output from cultured thalassemic cluster of differentiation 34 positive (CD34+) cells [54]. However, there are safety concerns with viral vectors, such as insertional mutagenesis, immune responses, and the potential for off-target effects, particularly in the context of in vivo delivery of lentivirus or transduction of autologous stem cells ex vivo. Non-viral approaches, such as the use of lipid nanoparticles or polymer-based systems, can improve safety by reducing the risk of insertional mutagenesis and immunogenicity, while potentially offering more precise delivery and better control overdosing [55]. Synthetic miR-15a sponges transfected into K562 cells upregulated miRNA target genes [56]. Chemically modified anti-miR-221 sponges formulated into nanoparticles showed efficient uptake into thalassemic erythroblasts, inhibiting miR-221 and its downstream effects [57].
Challenges for miRNA sponges include off-target effects, specificity, and developing means to achieve stable expression at effective doses. Rational design and testing of multi-targeting sponges based on miRNA expression patterns may improve potency. Recently, a screening system was developed to design and optimize inhibition of erythropoiesis-modulating miRNAs using lentiviral sponge libraries [58, 59]. Further innovation in delivery materials and testing in vivo will enable translation of miRNA sponges towards clinical applications.
Studies informed that miRNA sponges represent a promising technology for simultaneous inhibition of multiple pathogenic miRNAs in β-thalassemia. Advances in bioengineering design and delivery strategies are helping realize their therapeutic potential. With further optimization, miRNA sponges may provide an effective platform for rebalancing dysregulated miRNA expression and improving erythropoiesis in thalassemia patients.
Modulating miRNA processing enzymes
Rather than targeting individual miRNAs, an alternative approach is to modulate the upstream enzymes involved in miRNA biogenesis and processing. Key enzymes include Drosha and Dicer, which cleave and process miRNA precursors into their mature form. Early studies found Dicer knockdown increased HbF in K562 cells [60]. A high-throughput screen identified compounds that inhibit Dicer and promote HbF. The small molecule enoxacin was found to enhance γ-globin expression in erythroid cells by binding Dicer [61, 62].
However, directly inhibiting miRNA processing may have unintended effects. More recent work has focused on indirect modulation and targeting specific pathways. Metformin was shown to selectively impair Dicer processing of miR-15a/16-1 by reducing Dicer binding to XPO5, ameliorating thalassemic erythroblast dysfunction [63]. The flavonoid luteolin was found to downregulate Drosha expression through miR-22, leading to derepression of miR-15a/16-1 targets and increased erythroid output from thalassemic CD34+ cells [64].
While modulation of miRNA processing shows promise, predicting relevant functional effects remains challenging given the pleiotropic roles of these enzymes. Advances in understanding specificity determinants and off-target binding continue to emerge. For example, structure-guided engineering of XPO5 was recently described, which enabled selective inhibition of miR-15a/16-1 processing [65]. Ongoing research should uncover additional strategies for precise functional manipulation of miRNA biogenesis.
Further mechanistic and safety studies are needed to validate targeting of miRNA processing components for therapy. However, this approach presents opportunities for global manipulation of miRNA profiles relevant to thalassemia through small molecule, protein, or gene therapy-based strategies. Overall, the principle concept of the use of miRNA in thalassemia therapy is depicted in Figure 3.
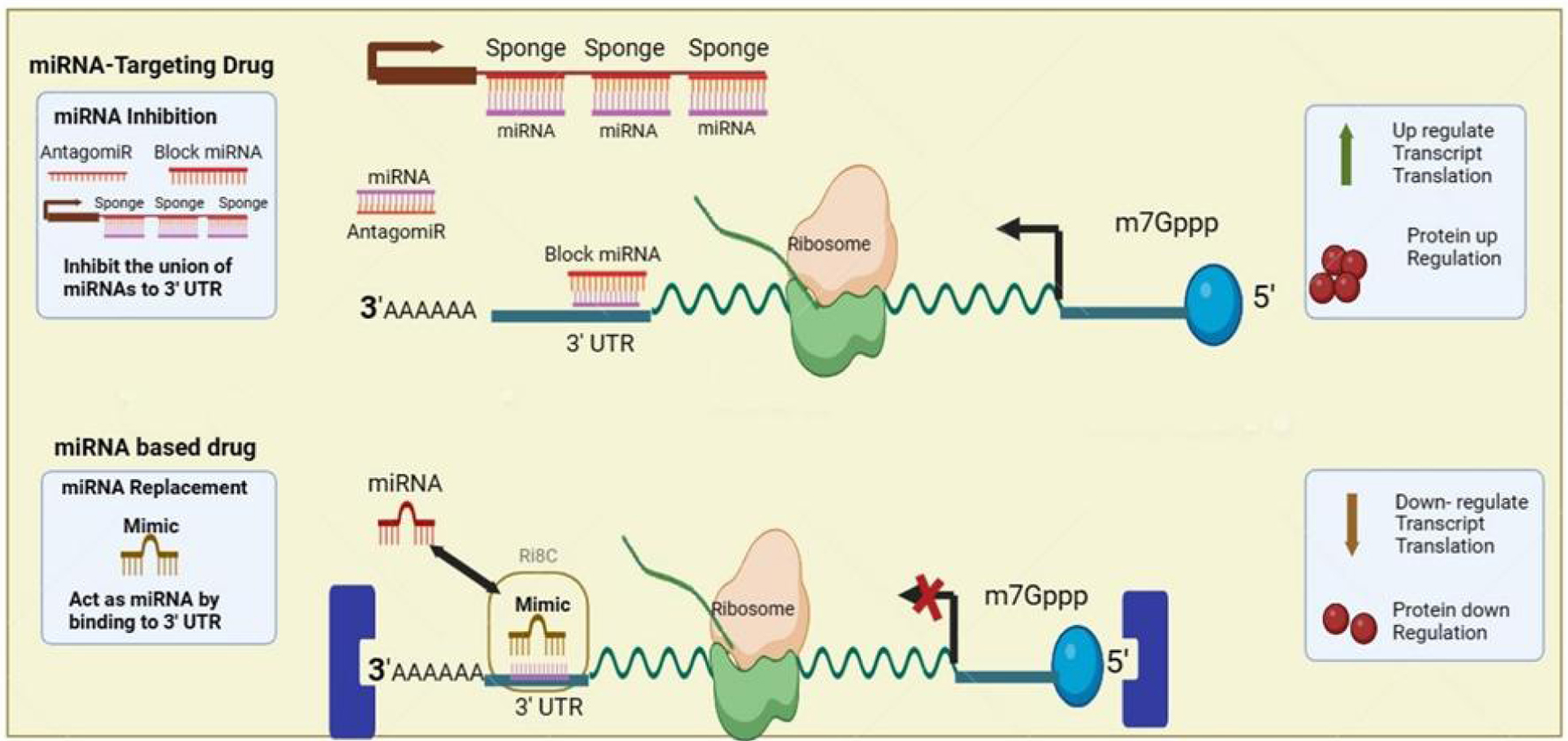 Click for large image |
Figure 3. MicroRNA (miRNA) application for thalassemia gene therapy. AntagomiRs function to inhibit miRNAs by binding and sequestering them, thus blocking their interaction with the 3' untranslated region (UTR) of messenger RNAs (mRNAs), which lifts the miRNA-mediated gene silencing and results in enhanced translation and protein production. MiRNA-based drugs utilize synthetic miRNA mimics that bind to the 3' UTR of target mRNAs to emulate natural miRNA functions, resulting in decreased mRNA translation and subsequent downregulation of protein synthesis. |
| Delivery Systems for MiRNA Therapeutics | ▴Top |
Efficient delivery poses a major challenge for clinical translation of miRNA-based drugs. Both miRNA mimics and inhibitors require optimized carriers to protect from degradation and enable uptake into target cells after systemic administration. A range of delivery platforms are being explored for thalassemia therapy, including both viral vectors and non-viral nanoparticles.
Lentiviral vectors can stably express miRNA mimics or sponges in erythroid cells, as demonstrated by testing in thalassemic mouse models and human CD34+ cells [14]. However, safety concerns exist regarding viral integration and immunogenicity. Non-viral options such as cationic lipoplexes and polyplexes can encapsulate therapeutic miRNAs but often suffer from toxicity, low efficiency, and rapid clearance. For example, polymeric nanoparticles loaded with anti-miR-221 showed low uptake in erythroid cells despite exhibiting high transfection in other cell types [66].
Recent strategies aim to enhance targeting and delivery by decorating nanoparticles with functional moieties like transferrin receptors. Transferrin-conjugated peptide nucleic acids targeting miR-15a exhibited improved cellular uptake and biological effects in thalassemic K562 cells. Platelet membrane-coated nanoparticles were recently shown to increase bone marrow delivery of miRNA mimics after intravenous injection in mice. Further testing of targeted delivery systems in vivo will facilitate clinical translation [67].
Overall, developing safe and effective carriers remains key for realizing clinical potential of miRNA therapeutics. Multifunctional combinatorial approaches leveraging diverse targeting, stealth, and endosomolytic strategies may help overcome current limitations in delivery to thalassemic erythroid cells.
| Preclinical Studies Testing MiRNA Therapies | ▴Top |
Mouse model studies
While in vitro models are valuable for initial screening, mouse models that closely recapitulate human thalassemia pathobiology are critical for evaluating miRNA therapies in vivo. Several murine models have been generated through genetic knockout or mutation of globin genes. The th3/+ severe thalassemic mouse model has been frequently used to test miRNA approaches.
Lentiviral vectors expressing miR-15a/16-1 mimics injected into th3/+ mice increased survival and improved anemia, with elevated erythrocyte counts and hemoglobin levels. Th3/+ mice treated with peptide-conjugated anti-miR-15a/16-1 inhibitor nanoparticles showed reduced spleen enlargement and increased hematocrit percentages. Addition of an immunomodulatory sequence abrogated inflammatory responses against the miRNA inhibitor nanoparticles in mice [68, 69].
Xenograft models by transplanting human thalassemic hematopoietic stem and progenitor cells into immunodeficient mice also enable testing miRNA therapies in a humanized system. Expression of a miR-15a sponge construct in CD34+ cells from thalassemia major patients prior to xenotransplantation improved engraftment and erythropoiesis in recipient mice [70].
While not fully recapitulating human physiology, mouse models allow crucial evaluation of pharmacokinetics, toxicity, and activity of miRNA therapies in vivo. Testing efficacy in diverse thalassemic mouse strains continues to build confidence for clinical translation. Further exploration of optimal delivery methods, dosing regimens, and combination treatments is warranted to support miRNA therapies moving into human trials.
Considerations for clinical translation
While ongoing preclinical research continues to demonstrate proof-of-concept for miRNA therapies in thalassemia, several key considerations remain to be addressed to enable clinical translation. Careful toxicity screening is critical, as some miRNA mimics or inhibitors have shown adverse effects on erythroid differentiation at high doses in vitro [71]. MiRNA-based therapies and short interfering RNA (siRNA) therapeutics share some similarities in their mechanisms of action and potential side effect profiles. Both can induce immune responses and off-target effects, although the extent and severity can vary depending on the delivery systems and specific targets involved. SiRNA therapeutics, for example Patisiran, an siRNA therapeutic for hereditary transthyretin-mediated amyloidosis, has demonstrated efficacy but is associated with adverse events such as peripheral edema and infusion-related reactions. Similar side effects may be anticipated with miRNA-based therapies, necessitating comprehensive safety evaluations [72, 73]. Demonstrating an adequate therapeutic index will be important. Pharmacokinetic studies in animal models are also essential to define systemic bioavailability, clearance, and biodistribution of miRNA drug candidates. This can help guide design of feasible dosing regimens [74].
Efficient and nontoxic delivery systems tailored for target cell populations remain a major challenge. While viral vectors show promise, concerns with immunogenicity, mutagenesis, and cost may limit clinical adoption. Non-viral delivery systems will require further innovation to achieve specific biodistribution to erythroid tissues, effective endosomal escape, and high-level cellular uptake [75]. Combining delivery technologies like ligand-mediated targeting with chemically modified miRNA mimics or inhibitors may provide synergistic solutions [33]. Definition of clinical endpoints and biomarkers to demonstrate activity and monitor effects must also be established. Potentially useful metrics include transfusion requirements, hemoglobin levels, reticulocyte counts, spleen size, and HbF levels.
Clinical translation will require demonstration of safety, efficacy, and reliable manufacturing and delivery that improves disease phenotypes and justifies cost. Priorities include optimizing miRNA drug potency and stability, minimizing off-target effects, defining biodistribution, establishing dosing regimens, and selecting viable delivery platforms. While challenges remain, the wealth of preclinical data supports the promising potential of miRNA therapies to progress into human clinical trials and provide transformative new treatment options for thalassemia patients. Ongoing research and development efforts will ultimately determine the viability and positioning of miRNA-based approaches in the evolving therapeutic landscape [76].
| Clinical Trials of MiRNA Therapies | ▴Top |
Current status and summary of early phase trials
While no miRNA-based therapies have yet advanced to clinical testing specifically for β-thalassemia, important lessons and precedents are being established through trials for other indications that can inform future development. The most advanced clinical program is the anti-miR-122 inhibitor miravirsen for treatment of hepatitis C virus (HCV) infection. Miravirsen showed dose-dependent reductions in HCV RNA levels in a phase 1 study, though efficacy was modest. A phase 2 trial demonstrated prolonged miRNA target suppression and antiviral activity with miravirsen monotherapy over 12 weeks. However, no further recent trials have been reported likely due to the advent of highly effective HCV direct-acting antiviral drugs [77].
The miR-29 mimic remomir was evaluated in early phase trials for reducing skin fibrosis and scarring with some promising results on safety, tolerability, and biomarker modulation, though efficacy was limited possibly due to suboptimal delivery [78]. Several other miRNA mimics or antimiRs are in preclinical development or early trials for indications like cancer but have yet to progress for hematological diseases like thalassemia. These early experiences highlight challenges with efficacy, delivery, and durability that remain to be solved. However, lessons in design, formulation, dosing, and safety will be informative for future thalassemia trials. Certainly, progress in delivery platforms and combinatorial approaches with gene or cell therapy may help overcome current limitations. While the clinical outlook is nascent, initial trials provide a glimpse towards realizing the therapeutic potential of miRNA-based medicines.
Safety and ethical considerations of miRNA technology in pediatric thalassemia therapy
The use of miRNA technology in pediatric thalassemia therapy presents both promising opportunities and significant challenges. MiRNAs are crucial in regulating gene expression during development, so their modulation in children requires careful assessment of potential impacts on growth and development. Preclinical studies must rigorously evaluate these impacts to ensure safety [79]. Additionally, the long-term effects of miRNA therapies are not fully understood, and children may require lifelong management of thalassemia. Long-term follow-up studies are essential to monitor for delayed adverse effects, ensuring sustained safety and efficacy. The developing immune systems of pediatric patients may also react differently to miRNA therapies compared to adults, necessitating delivery systems that minimize immune activation and enhance biocompatibility [80].
Ethical challenges are equally important. Obtaining informed consent is complex, as parents or guardians must make decisions on behalf of the child. It is imperative they are fully informed about the potential risks and benefits of miRNA therapies [81]. Balancing the risks and benefits is crucial, adhering to the ethical principle of “do no harm”. Pediatric therapies must prioritize the child’s best interests, with rigorous risk-benefit assessments guiding decision-making. Ensuring equitable access to these innovative treatments is also an ethical imperative, regardless of socio-economic status [82]. Addressing these safety and ethical challenges requires a collaborative approach involving clinicians, researchers, ethicists, and regulatory bodies to establish guidelines and protocols that prioritize the safety and well-being of pediatric patients.
| Conclusion | ▴Top |
Summary of current state and future outlook
In summary, β-thalassemia represents a challenging hematological disease where innovative therapeutic approaches are needed to move beyond the limitations of chronic transfusions and iron chelation. Modulation of miRNAs has rapidly emerged as a promising strategy based on their critical regulatory roles in erythropoiesis and thalassemic pathophysiology. While only in the early stages of development, preclinical studies have already demonstrated the ability of miRNA mimics, inhibitors, and sponges to ameliorate disease phenotypes by targeting key miRNAs dysregulated in thalassemia. This has laid the groundwork for eventual clinical translation. However, work remains to optimize delivery systems, improve in vivo efficacy and safety, develop combinatorial approaches with genetic therapies, and establish feasible manufacturing scale-up.
If challenges in stability, biodistribution, and targeted delivery can be solved, miRNA-based medicines have immense potential to expand and transform treatment options for thalassemia in the future. Ongoing innovation in the field continues to generate excitement as more becomes understood regarding the complex biology of miRNAs in hematopoiesis and their therapeutic manipulation. While the clinical outlook remains nascent, creative solutions emerging from integrating fields like materials science, nanotechnology, and gene therapy may accelerate realizing the promise of miRNAs for thalassemia patients.
Potential for miRNAs to transform care for thalassemia patients
Modulating dysregulated miRNAs offers the possibility to precisely correct molecular mechanism underlying thalassemia. MiRNAs can potentially overcome limitations of current management by protecting patients from ineffective erythropoiesis and anemia. Combining miRNA therapies with curative genetic approaches may provide synergistic solutions improving accessibility and outcomes. If advanced in economically viable and scalable ways, miRNA medicines could make transformative impacts expanding treatment options and radically improving quality of life for thalassemia patients worldwide.
Acknowledgments
We thank Wuri, the librarian, for assistance in searching for articles and resources for this publication.
Financial Disclosure
Ministry of Education, Culture, Research, and Technology Republic of Indonesia, Grants number 27.49/UN23.37/PT.01.03/II/2023 for International Research Collaboration (IRC) grant of Universitas Jenderal Soedirman.
Conflict of Interest
The authors declare no conflict of interest in the development and submission of this manuscript.
Author Contributions
LR: principal conceptualization, initial manuscript development, and final manuscript review; TW: manuscript development and image creation; WS and IMN: manuscript development; THS: manuscript development and critical review.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
miRNA: microRNA; HbF: fetal hemoglobin; UTR: untranslated region; mRNA: messenger RNA; RNAi: RNA interference; CD34+: cluster of differentiation 34 positive; CRISPR/Cas9: clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9; HCV: hepatitis C virus; RNA: ribonucleic acid; PCR: polymerase chain reaction; PLGA: poly-lactic-co-glycolic acid; DGCR8: DiGeorge syndrome critical region 8; XPO5: exportin-5; USP36: ubiquitin-specific peptidase 36; ULK1: unc-51 like autophagy activating kinase 1; LKB1: liver kinase B1; AMPK: AMP-activated protein kinase; mTOR: mammalian target of rapamycin; BCL2: B-cell lymphoma 2; NRF2: nuclear factor erythroid 2-related factor 2; GATA1: GATA-binding protein 1; Tal1: T-cell acute lymphocytic leukemia protein 1; KLF8: Kruppel-like factor 8; MAPK: mitogen-activated protein kinase; MYB: Myb proto-oncogene, transcription factor; KLF3: Kruppel-like factor 3; BCL11A: B-cell lymphoma/leukemia 11A; SOX6: SRY-box transcription factor 6; EKLF: erythroid Kruppel-like factor
| References | ▴Top |
- Angastiniotis M, Lobitz S. Thalassemias: an overview.
Int J Neonatal Screen. 2019;5(1):16.
doi pubmed pmc - Lee JS, Rhee TM, Jeon K, Cho Y, Lee SW, Han KD, Seong MW, et
al. Epidemiologic trends of Thalassemia, 2006-2018: a nationwide population-based study. J Clin
Med. 2022;11(9):2289.
doi pubmed pmc - Susanto Z, Siswandari W, Rujito L. Cd60 (GTG>GAG)/Hb
Cagliari mutation was found in scanning of beta-thalassemia alleles from patients of East
Kalimantan, Indonesia. Mol Genet Metab Rep. 2020;22:100550.
doi pubmed pmc - Kattamis A, Forni GL, Aydinok Y, Viprakasit V. Changing
patterns in the epidemiology of beta-thalassemia. Eur J Haematol.
2020;105(6):692-703.
doi pubmed pmc - Hokland P, Daar S, Khair W, Sheth S, Taher AT, Torti L,
Hantaweepant C, et al. Thalassaemia-A global view. Br J Haematol.
2023;201(2):199-214.
doi pubmed - Farmakis D, Porter J, Taher A, Domenica Cappellini M,
Angastiniotis M, Eleftheriou A. 2021 Thalassaemia International Federation Guidelines for the
Management of Transfusion-dependent Thalassemia. Hemasphere. 2022;6(8):e732.
doi pubmed pmc - An R, Avanaki A, Thota P, Nemade S, Mehta A, Gurkan UA.
Point-of-care diagnostic test for beta-thalassemia. Biosensors (Basel). 2024;14(2):83.
doi pubmed pmc - Xu JZ, Foe M, Tanongsaksakul W, Suksangpleng T, Ekwattanakit
S, Riolueang S, Telen MJ, et al. Identification of optimal thalassemia screening strategies for
migrant populations in Thailand using a qualitative approach. BMC Public Health.
2021;21(1):1796.
doi pubmed pmc - Mustafa I, Firdous N, Shebl FM, Shi Z, Saeed M, Zahir Z,
Zayed H. Genetic epidemiology of beta-thalassemia in the Maldives: 23 years of a
beta-thalassemia screening program. Gene. 2020;741:144544.
doi pubmed - De Simone G, Quattrocchi A, Mancini B, di Masi A, Nervi C,
Ascenzi P. Thalassemias: from gene to therapy. Mol Aspects Med. 2022;84:101028.
doi pubmed - Pilo F, Angelucci E. Luspatercept to treat beta-thalassemia.
Drugs Today (Barc). 2020;56(7):447-458.
doi pubmed - Musallam KM, Sheth S, Cappellini MD, Kattamis A, Kuo KHM,
Taher AT. Luspatercept for transfusion-dependent beta-thalassemia: time to get real. Ther Adv
Hematol. 2023;14:20406207231195594.
doi pubmed pmc - Fellmann C, Gowen BG, Lin PC, Doudna JA, Corn JE.
Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov.
2017;16(2):89-100.
doi pubmed pmc - Nath A, Rayabaram J, Ijee S, Bagchi A, Chaudhury AD, Roy D,
Chambayil K, et al. Comprehensive analysis of microRNAs in human adult erythropoiesis. Cells.
2021;10(11):3018.
doi pubmed pmc - Philippidis A. CASGEVY makes history as FDA approves first
CRISPR/Cas9 genome edited therapy. Hum Gene Ther. 2024;35(1-2):1-4.
doi pubmed - Brusson M, Miccio A. Genome editing approaches to
beta-hemoglobinopathies. Prog Mol Biol Transl Sci. 2021;182:153-183.
doi pubmed - Verma HK, Ratre YK, Bhaskar L, Colombatti R. Erythrocyte
microRNAs: a tiny magic bullet with great potential for sickle cell disease therapy. Ann
Hematol. 2021;100(3):607-614.
doi pubmed - Katsantoni E. Omics studies in hemoglobinopathies. Mol Diagn
Ther. 2019;23(2):223-234.
doi pubmed - Martinez I, Hayes KE, Barr JA, Harold AD, Xie M, Bukhari
SIA, Vasudevan S, et al. An Exportin-1-dependent microRNA biogenesis pathway during human cell
quiescence. Proc Natl Acad Sci U S A. 2017;114(25):E4961-E4970.
doi pubmed pmc - Wu K, He J, Pu W, Peng Y. The role of Exportin-5 in MicroRNA
biogenesis and cancer. Genomics Proteomics Bioinformatics. 2018;16(2):120-126.
doi pubmed pmc - Li Y, Carey TS, Feng CH, Zhu HM, Sun XX, Dai MS. The
Ubiquitin-specific protease USP36 associates with the microprocessor complex and regulates miRNA
biogenesis by SUMOylating DGCR8. Cancer Res Commun. 2023;3(3):459-470.
doi pubmed pmc - Dang TL, Le CT, Le MN, Nguyen TD, Nguyen TL, Bao S, Li S, et
al. Select amino acids in DGCR8 are essential for the UGU-pri-miRNA interaction and processing.
Commun Biol. 2020;3(1):344.
doi pubmed pmc - Sewastianik T, Straubhaar JR, Zhao JJ, Samur MK, Adler K,
Tanton HE, Shanmugam V, et al. miR-15a/16-1 deletion in activated B cells promotes plasma cell
and mature B-cell neoplasms. Blood. 2021;137(14):1905-1919.
doi pubmed pmc - Origa R. beta-Thalassemia. Genet Med.
2017;19(6):609-619.
doi pubmed - Ali H, Khan F. Non-coding RNA: an emerging modulator of β-globin regulation and β-hemoglobinopathies. Mol Med Commun. 2022;2:173-189
- Wang F, Ling L, Yu D. MicroRNAs in beta-thalassemia.
Am J Med Sci. 2021;362(1):5-12.
doi pubmed - Cazzola M. Ineffective erythropoiesis and its treatment.
Blood. 2022;139(16):2460-2470.
doi pubmed - Oikonomidou PR, Rivella S. What can we learn from
ineffective erythropoiesis in thalassemia? Blood Rev. 2018;32(2):130-143.
doi pubmed pmc - Pekarsky Y, Balatti V, Croce CM. BCL2 and miR-15/16: from
gene discovery to treatment. Cell Death Differ. 2018;25(1):21-26.
doi pubmed pmc - Li B, Zhu X, Ward CM, Starlard-Davenport A, Takezaki M,
Berry A, Ward A, et al. MIR-144-mediated NRF2 gene silencing inhibits fetal hemoglobin
expression in sickle cell disease. Exp Hematol. 2019;70:85-96.e85.
doi pubmed pmc - Cyrus C. The Role of miRNAs as Therapeutic tools in sickle
cell disease. Medicina (Kaunas). 2021;57(10):1106.
doi pubmed pmc - Wang D, Si S, Wang Q, Luo G, Du Q, Liang Q, Guo X, et al.
MiR-27a promotes hemin-induced erythroid differentiation of K562 cells by targeting CDC25B. Cell
Physiol Biochem. 2018;46(1):365-374.
doi pubmed - Hildebrand D, Eberle ME, Wolfle SM, Egler F, Sahin D, Sahr
A, Bode KA, et al. Hsa-miR-99b/let-7e/miR-125a cluster regulates pathogen recognition
receptor-stimulated suppressive antigen-presenting cells. Front Immunol. 2018;9:1224.
doi pubmed pmc - Morris VA, Zhang A, Yang T, Stirewalt DL, Ramamurthy R,
Meshinchi S, Oehler VG. MicroRNA-150 expression induces myeloid differentiation of human acute
leukemia cells and normal hematopoietic progenitors. PLoS One. 2013;8(9):e75815.
doi pubmed pmc - Katsaraki K, Karousi P, Artemaki PI, Scorilas A, Pappa V,
Kontos CK, Papageorgiou SG. MicroRNAs: tiny regulators of gene expression with pivotal roles in
normal B-cell development and B-cell chronic lymphocytic leukemia. Cancers (Basel).
2021;13(4):593.
doi pubmed pmc - Sankaran VG, Orkin SH. The switch from fetal to adult
hemoglobin. Cold Spring Harb Perspect Med. 2013;3(1):a011643.
doi pubmed pmc - Saki N, Abroun S, Soleimani M, Kavianpour M, Shahjahani M,
Mohammadi-Asl J, Hajizamani S. MicroRNA expression in beta-Thalassemia and sickle cell disease:
A Role in The Induction of Fetal Hemoglobin. Cell J. 2016;17(4):583-592.
doi pubmed pmc - Kim M, Civin CI, Kingsbury TJ. MicroRNAs as regulators and
effectors of hematopoietic transcription factors. Wiley Interdiscip Rev RNA.
2019;10(5):e1537.
doi pubmed - Starlard-Davenport A, Gu Q, Pace BS. Targeting genetic
modifiers of HBG gene expression in sickle cell disease: the miRNA option. Mol Diagn Ther.
2022;26(5):497-509.
doi pubmed pmc - Sun KT, Huang YN, Palanisamy K, Chang SS, Wang IK, Wu KH,
Chen P, et al. Reciprocal regulation of gamma-globin expression by exo-miRNAs: Relevance to
gamma-globin silencing in beta-thalassemia major. Sci Rep. 2017;7(1):202.
doi pubmed pmc - Kargutkar N, Sawant-Mulay M, Hariharan P, Chandrakala S,
Nadkarni A. Role of microRNA in hydroxyurea mediated HbF induction in sickle cell anaemia
patients. Sci Rep. 2023;13(1):369.
doi pubmed pmc - d'Arqom A. Nucleic Acid Therapy for beta-Thalassemia.
Biologics. 2020;14:95-105.
doi pubmed pmc - Levin C, Koren A, Rebibo-Sabbah A, Levin M, Koifman N,
Brenner B, Aharon A. Extracellular vesicle MicroRNA that are involved in beta-Thalassemia
complications. Int J Mol Sci. 2021;22(18):9760.
doi pubmed pmc - Arghiani N, Shah K. Modulating microRNAs in cancer:
next-generation therapies. Cancer Biol Med. 2021;19(3):289-304.
doi pubmed pmc - Undi RB, Kandi R, Gutti RK. MicroRNAs as haematopoiesis
regulators. Adv Hematol. 2013;2013:695754.
doi pubmed pmc - Kim JY, Jung EJ, Kim JM, Son Y, Lee HS, Kwag SJ, Park JH, et
al. MiR-221 and miR-222 regulate cell cycle progression and affect chemosensitivity in breast
cancer by targeting ANXA3. Exp Ther Med. 2023;25(3):127.
doi pubmed pmc - Han H, Bartolo R, Li J, Shahbazi MA, Santos HA. Biomimetic
platelet membrane-coated nanoparticles for targeted therapy. Eur J Pharm Biopharm.
2022;172:1-15.
doi pubmed - Fang X, Shen F, Lechauve C, Xu P, Zhao G, Itkow J, Wu F, et
al. miR-144/451 represses the LKB1/AMPK/mTOR pathway to promote red cell precursor survival
during recovery from acute anemia. Haematologica. 2018;103(3):406-416.
doi pubmed pmc - Sangokoya C, Telen MJ, Chi JT. microRNA miR-144 modulates
oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood.
2010;116(20):4338-4348.
doi pubmed pmc - Keith J, Christakopoulos GE, Fernandez AG, Yao Y, Zhang J,
Mayberry K, Telange R, et al. Loss of miR-144/451 alleviates beta-thalassemia by stimulating
ULK1-mediated autophagy of free alpha-globin. Blood. 2023;142(10):918-932.
doi pubmed pmc - Ebert MS, Sharp PA. MicroRNA sponges: progress and
possibilities. RNA. 2010;16(11):2043-2050.
doi pubmed pmc - Ling L, Wang F, Li Y, He S, Wu F, Yang L, Xu L, et al.
Depletion of miR-144/451 alleviates anemia in beta-thalassemic mice. Blood Adv.
2024;8(10):2565-2570.
doi pubmed pmc - Athanasopoulou K, Chondrou V, Xiropotamos P, Psarias G,
Vasilopoulos Y, Georgakilas GK, Sgourou A. Transcriptional repression of lncRNA and miRNA
subsets mediated by LRF during erythropoiesis. J Mol Med (Berl).
2023;101(9):1097-1112.
doi pubmed pmc - Felli N, Fontana L, Pelosi E, Botta R, Bonci D, Facchiano F,
Liuzzi F, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell
growth via kit receptor down-modulation. Proc Natl Acad Sci U S A.
2005;102(50):18081-18086.
doi pubmed pmc - Schilb AL, Scheidt JH, Vaidya AM, Sun Z, Sun D, Lee S, Lu
ZR. Optimization of synthesis of the amino lipid ECO for effective delivery of nucleic acids.
Pharmaceuticals (Basel). 2021;14(10):1016.
doi pubmed pmc - Anelli L, Zagaria A, Specchia G, Musto P, Albano F.
Dysregulation of miRNA in Leukemia: exploiting miRNA expression profiles as biomarkers. Int J
Mol Sci. 2021;22(13):7156.
doi pubmed pmc - Sevcikova A, Fridrichova I, Nikolaieva N, Kalinkova L,
Omelka R, Martiniakova M, Ciernikova S. Clinical significance of micrornas in hematologic
malignancies and hematopoietic stem cell transplantation. Cancers (Basel).
2023;15(9):2658.
doi pubmed pmc - Papapetrou EP, Korkola JE, Sadelain M. A genetic strategy
for single and combinatorial analysis of miRNA function in mammalian hematopoietic stem cells.
Stem Cells. 2010;28(2):287-296.
doi pubmed - Chen L, Zhang K, Shi Z, Zhang A, Jia Z, Wang G, Pu P, et al.
A lentivirus-mediated miR-23b sponge diminishes the malignant phenotype of glioma cells in vitro
and in vivo. Oncol Rep. 2014;31(4):1573-1580.
doi pubmed - Bianchi N, Zuccato C, Finotti A, Lampronti I, Borgatti M,
Gambari R. Involvement of miRNA in erythroid differentiation. Epigenomics.
2012;4(1):51-65.
doi pubmed - Amaya M, Desai M, Gnanapragasam MN, Wang SZ, Zu Zhu S,
Williams DC, Jr., Ginder GD. Mi2beta-mediated silencing of the fetal gamma-globin gene in adult
erythroid cells. Blood. 2013;121(17):3493-3501.
doi pubmed pmc - Ahmadi A, Moradi S. In silico analysis suggests the
RNAi-enhancing antibiotic enoxacin as a potential inhibitor of SARS-CoV-2 infection. Sci Rep.
2021;11(1):10271.
doi pubmed pmc - Pedroza-Torres A, Romero-Cordoba SL, Justo-Garrido M,
Salido-Guadarrama I, Rodriguez-Bautista R, Montano S, Muniz-Mendoza R, et al. MicroRNAs in tumor
cell metabolism: roles and therapeutic opportunities. Front Oncol. 2019;9:1404.
doi pubmed pmc - Ahmed F, Ijaz B, Ahmad Z, Farooq N, Sarwar MB, Husnain T.
Modification of miRNA Expression through plant extracts and compounds against breast cancer:
Mechanism and translational significance. Phytomedicine. 2020;68:153168.
doi pubmed - Bayraktar E, Bayraktar R, Oztatlici H, Lopez-Berestein G,
Amero P, Rodriguez-Aguayo C. Targeting miRNAs and other non-coding RNAs as a therapeutic
approach: an update. Noncoding RNA. 2023;9(2):27.
doi pubmed pmc - Volpi S, Cancelli U, Neri M, Corradini R. Multifunctional
delivery systems for peptide nucleic acids. Pharmaceuticals (Basel). 2020;14(1):14.
doi pubmed pmc - Gambari R. Chapter 39 - delivery and biological activity of therapeutic miRNAs and miRNA modifiers. In: Sen CK (ed). MicroRNA in Regenerative Medicine. Oxford: Academic Press. 2015:1017-1048.
- Kautz L, Jung G, Du X, Gabayan V, Chapman J, Nasoff M,
Nemeth E, et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse
model of beta-thalassemia. Blood. 2015;126(17):2031-2037.
doi pubmed pmc - Mehta M, Satija S, Paudel KR, Malyla V, Kannaujiya VK,
Chellappan DK, Bebawy M, et al. Targeting respiratory diseases using miRNA inhibitor based
nanotherapeutics: Current status and future perspectives. Nanomedicine. 2021;31:102303.
doi pubmed - Jia W, Jia S, Chen P, He Y. Construction and Analysis of a
Long Non-Coding RNA (lncRNA)-associated ceRNA Network in beta-Thalassemia and hereditary
persistence of fetal hemoglobin. Med Sci Monit. 2019;25:7079-7086.
doi pubmed pmc - Xu L, Wu F, Yang L, Wang F, Zhang T, Deng X, Zhang X, et al.
miR-144/451 inhibits c-Myc to promote erythroid differentiation. FASEB J.
2020;34(10):13194-13210.
doi pubmed - Adams D, Gonzalez-Duarte A, O'Riordan WD, Yang CC, Ueda M,
Kristen AV, Tournev I, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin
amyloidosis. N Engl J Med. 2018;379(1):11-21.
doi pubmed - Yamashita T, Ueda M, Koike H, et al. Patisiran, an RNAi therapeutic for patients with hereditary transthyretin-mediated amyloidosis: Sub-analysis in Japanese patients from the APOLLO study. Neurol Clin Neurosci. 2020;8:251-260.
- Herkt M, Thum T. Pharmacokinetics and proceedings in
clinical application of nucleic acid therapeutics. Mol Ther. 2021;29(2):521-539.
doi pubmed pmc - Sinclair F, Begum AA, Dai CC, Toth I, Moyle PM. Recent
advances in the delivery and applications of nonviral CRISPR/Cas9 gene editing. Drug Deliv
Transl Res. 2023;13(5):1500-1519.
doi pubmed pmc - Kotowska-Zimmer A, Pewinska M, Olejniczak M. Artificial
miRNAs as therapeutic tools: Challenges and opportunities. Wiley Interdiscip Rev RNA.
2021;12(4):e1640.
doi pubmed - van der Ree MH, van der Meer AJ, van Nuenen AC, de Bruijne
J, Ottosen S, Janssen HL, Kootstra NA, et al. Miravirsen dosing in chronic hepatitis C patients
results in decreased microRNA-122 levels without affecting other microRNAs in plasma. Aliment
Pharmacol Ther. 2016;43(1):102-113.
doi pubmed - Gallant-Behm CL, Piper J, Lynch JM, Seto AG, Hong SJ, Mustoe
TA, Maari C, et al. A MicroRNA-29 mimic (Remlarsen) represses extracellular matrix expression
and fibroplasia in the skin. J Invest Dermatol. 2019;139(5):1073-1081.
doi pubmed - Jeong HR, Hwang IT. MicroRNAs as novel biomarkers for the
diagnosis and treatment of pediatric diseases. Clin Exp Pediatr. 2024;67(3):119-125.
doi pubmed pmc - Seyhan AA. Trials and tribulations of MicroRNA therapeutics.
Int J Mol Sci. 2024;25(3):1469.
doi pubmed pmc - Iacomino G. miRNAs: the road from bench to bedside. Genes
(Basel). 2023;14(2):314.
doi pubmed pmc - Ormond KE, Blasimme A, Vayena E. Ethical aspects of
pediatric genetic care: testing and treatment. Pediatr Clin North Am.
2023;70(5):1029-1046.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Clinical Medicine Research is published by Elmer Press Inc.