| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website https://jocmr.elmerjournals.com |
Original Article
Volume 17, Number 3, March 2025, pages 153-163

Oral Pulmonary Arterial Hypertension-Targeted Therapy in Patients With Pulmonary Hypertension due to Interstitial Lung Disease
Kavitha C. Selvana, b, Krittika Teerapuncharoenc, Remzi Baga, d, e
aSection of Pulmonary and Critical Care Medicine, Department of Medicine, University
of Chicago Medical Center, Chicago, IL, USA
bCurrent: Division of Pulmonary and
critical Care Medicine, Department of Medicine, Northwestern University, Chicago, IL,
USA
cDivision of Respiratory Disease and Tuberculosis, Faculty of Medicine
Siriraj Hospital, Mahidol University, Bangkok, Thailand
dCurrent: Division of
Lung Failure and Transplant, Department of Transplantation, Mayo Clinic, Jacksonville, FL,
USA
eCorresponding Author: Remzi Bag, Mayo Clinic, Jacksonville, FL 32224, USA
Manuscript submitted November 13, 2024, accepted February 13, 2025, published online March 10,
2025
Short title: Oral PAH-Targeted Therapy in PH-ILD
doi:
https://doi.org/10.14740/jocmr6164
| Abstract | ▴Top |
Background: The aim of the study was to determine whether treatment with oral pulmonary arterial hypertension (PAH)-targeted therapy is associated with functional or hemodynamic improvement in patients with pulmonary hypertension due to interstitial lung disease (PH-ILD).
Methods: We conducted a retrospective review of 1,507 consented patients with pulmonary hypertension (PH) from the University of Chicago PH Registry. Exclusion criteria included: enrollment in PH-related clinical trials, use of inhaled treprostinil or iloprost and prior PAH-targeted therapy initiated before consenting to registry enrollment, thus precluding baseline data. Data analyzed included demographics, interstitial lung disease (ILD) classification, PAH-targeted therapy, functional data, hemodynamics, and N-terminal pro-B-type natriuretic peptide (NT-proBNP) before and after initiation of treatment. Data were analyzed using paired t-test, or related-samples Wilcoxon signed rank test.
Results: Of 37 patients included, 27 (73%) received treatment with one PAH-targeted therapy and nine (24%) received dual therapy. At baseline, median NT-proBNP was 1,498 ng/dL (675 - 3,208), mean pulmonary artery pressure (mPAP) was 45 ± 11 mm Hg, and pulmonary vascular resistance (PVR) of 9 ± 4 Wood units (WU). In patients with measurements both before and after treatment with PAH-targeted therapy, there was a decrease in PVR (n = 13, 8 vs. 5 WU, P < 0.001), an increase in cardiac output (n = 13, 4 vs. 5 L/min, P = 0.014), and a decrease in NT-proBNP levels (n = 26, 1,421 vs. 842 ng/dL, P = 0.045).
Conclusions: In this study, use of PAH-targeted therapy in patients with PH-ILD was associated with statistically significant and clinically meaningful improvements in NT-proBNP and pulmonary hemodynamics.
Keywords: Pulmonary hypertension; Interstitial lung disease; PAH-targeted therapy
| Introduction | ▴Top |
Pulmonary hypertension (PH) is a vascular disease defined by a mean pulmonary artery pressure (mPAP) > 20 mm Hg and a pulmonary vascular resistance (PVR) ≥ 2 Wood units (WU) on right heart catheterization (RHC), except in isolated post-capillary PH, in which the PVR criteria need not to be met [1]. The World Symposium on Pulmonary Hypertension (WSPH) has further subclassified PH based on etiology, with group 1 referring to those with pulmonary arterial hypertension (PAH), and group 3 referring to those with pre-capillary PH and hypoxia and/or chronic lung diseases including interstitial lung disease (ILD) [1].
Patients with ILD frequently develop PH (PH-ILD) as a consequence of the vascular and parenchymal remodeling that occurs during disease progression [2]. Prevalence varies widely based on ILD subtype, ranging from 18% to 50% overall, with the highest rates associated with idiopathic pulmonary fibrosis (IPF), connective tissue disease-associated ILD (CTD-ILD), combined pulmonary fibrosis and emphysema (CPFE), and pulmonary Langerhans cell histiocytosis (PLCH) [3-6]. In cohorts of patients with PH and all types of ILD, majority have mild PH, defined as a mPAP between 25 and 35 mm Hg on RHC [7, 8]. However, a small cohort evaluating RHC hemodynamics in patients with antisynthetase syndrome found that 13 of the 16 patients with pre-capillary PH by RHC had severe PH (mPAP ≥ 35 mm Hg) [9]. Additionally, between 2% and 10% of patients with IPF and PH have severe PH [10, 11]. The development of PH-ILD is associated with increased dyspnea, reduced lung function, lower 6-min walk distance (6MWD), increased oxygen requirement, and increased mortality [7, 12-14].
Food and Drug Administration (FDA) approval for treatment with PAH-targeted therapy has been limited to those patients with PAH and chronic thromboembolic PH (CTEPH). A recent study of inhaled treprostinil in patients with PH-ILD found improvement in 6MWD and reduction in pro-BNP in the treatment group and has led to excitement amongst PH providers who are now able to offer pharmacologic treatment to this subset of patients [15]. However, studies evaluating the efficacy of other PAH-targeted therapy in this patient population remain limited and controversial [16-26]. The objective of our study was to evaluate the functional and hemodynamic effect of other PAH-targeted therapy in the treatment of PH-ILD.
| Materials and Methods | ▴Top |
We conducted a retrospective chart review of 1,507 patients with written informed consent enrolled in the University of Chicago Medicine PH registry between January 1, 1997 and October 30, 2021. This study was approved by the Institutional Review Board of the University of Chicago Medical Center in Chicago, IL (IRB17-1427). The study was conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration. Entries were filtered by a registry ICD-9/10 diagnosis code of ILD or connective tissue disease (CTD) or the presence of restriction on pulmonary function test (PFT) (defined as total lung capacity (TLC) ≤ 70% predicted or forced vital capacity (FVC) ≤ 70% predicted). A second filter screening for the presence of any PH-targeted therapy (regardless of the rationale for the choice of agent selected) was then applied. The charts and chest computed tomography (CT) images of the remaining patients were then reviewed in a blinded fashion by two of three study authors (RB, KS), one of whom is a PH expert, to confirm a diagnosis of ILD (defined as the presence of reticulation, traction bronchiectasis and/or honeycombing on high-resolution computed tomography (HRCT) of the chest) and a diagnosis of WSPH group 3 PH, based on the 2018 definition [1]. Patients were excluded from analysis if they had minimal radiographic interstitial changes (interstitial lung abnormalities (ILAs)) without the presence of ILD (therefore classified as WSPH group 1 PH), a diagnosis of sarcoidosis, received treatment with inhaled treprostinil or iloprost, prior PAH-targeted therapy initiated before consenting to registry enrollment, thus precluding baseline data), or were concurrently enrolled in PH-related clinical trials (Fig. 1).
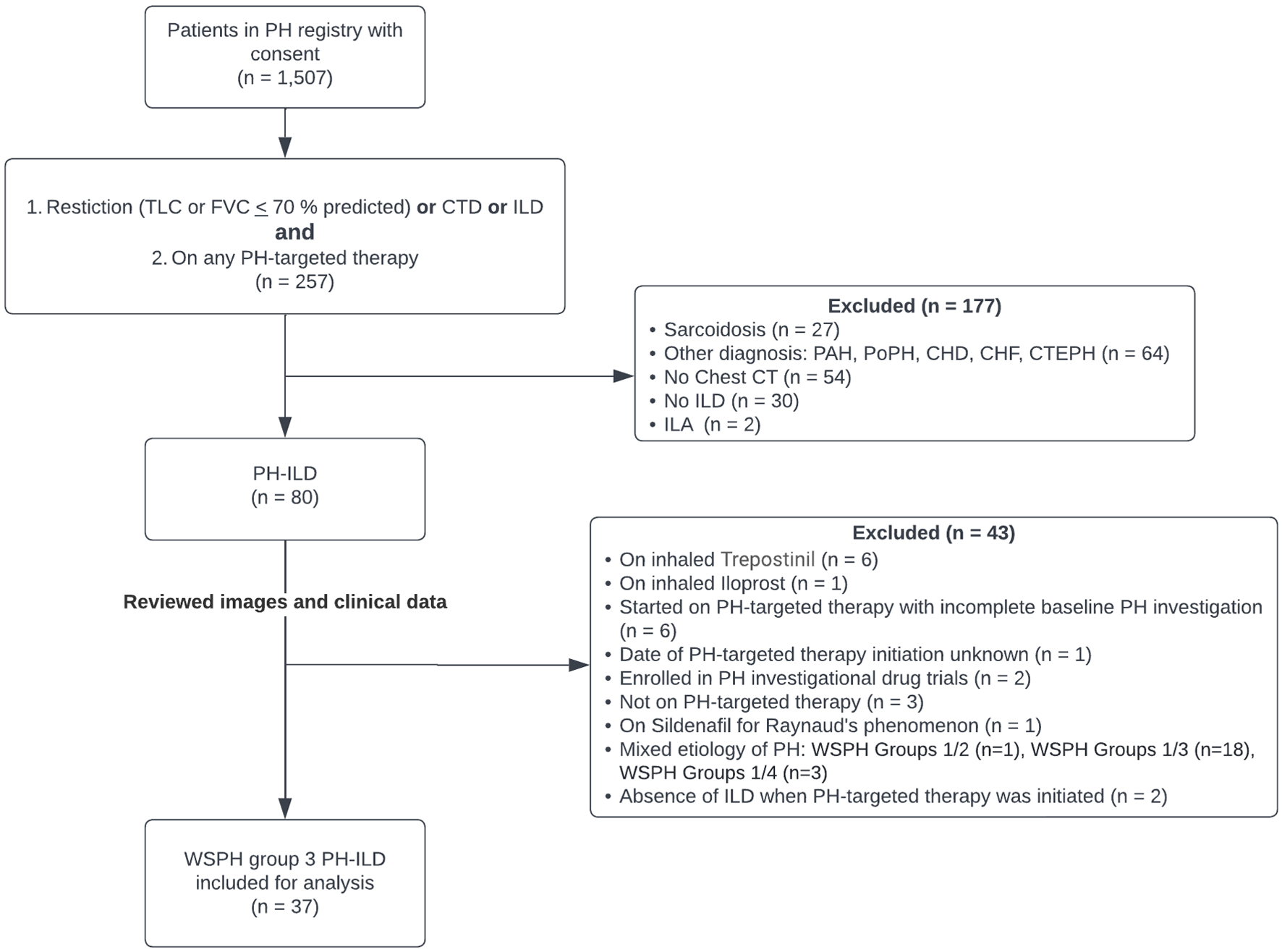 Click for large image |
Figure 1. Patient screening. CHD: congenital heart disease; CHF: congestive heart failure; CT: computed tomography; CTD: connective tissue disease; CTEPH: chronic thromboembolic pulmonary hypertension; FVC: forced vital capacity; ILA: interstitial lung abnormality; ILD: interstitial lung disease; PAH: pulmonary arterial hypertension; PH: pulmonary hypertension; PoPH: portopulmonary hypertension; TLC: total lung capacity; WSPH: World Symposium of Pulmonary Hypertension. |
Demographic information collected included age, race, and relevant comorbid conditions. The diagnosis of ILD was further characterized by subtype and radiographic pattern, as determined by two-physician agreement (RB, KS) with 100% concordance for all cases. Information regarding PAH-targeted therapy, including date of onset, PAH-targeted therapy class (phosphodiesterase-5 inhibitor (PDE5-I), endothelin receptor antagonist (ERA), soluble guanylyl cyclase stimulator (sGCS), and/or prostacyclin), dose, and duration of therapy were collected. Determination of functional status was assessed by review of World Health Organization (WHO) functional class (FC), 6MWD, and/or metabolic equivalents (METs) achieved on exercise stress test. Data from PFTs were collected. Hemodynamic data collected included right atrial pressure, right ventricular pressure, mPAP, pulmonary capillary wedge pressure (PCWP), PVR, cardiac output (CO), and cardiac index (CI) (thermodilution or Fick) as measured by RHC, and the presence of PH was defined as mPAP > 20 mm Hg and PVR ≥ 3 WU. Laboratory data including complete blood counts, creatinine, and NT-proBNP were collected. Data on functional status, hemodynamics, and laboratory studies were collected at baseline (prior to initiation of PAH-targeted therapy) and upon follow-up, within 14 to 90 weeks after initiation of treatment with PAH-targeted therapy.
Data are presented as means ± standard deviation (SD) or medians with interquartile range (IQR) when appropriate. Differences between groups were compared using paired t-test or related-samples Wilcoxon signed rank test. Survival analysis was performed using a Kaplan-Meier estimate.
| Results | ▴Top |
After initial screening, 37 patients were included. Characteristics at baseline for the entire cohort are summarized in Table 1. The average age was 60 ± 11 years and 78% of patients were female. Common comorbid conditions included CTD (n = 27, 73%), hypertension (n = 19, 52%), obesity (n = 15, 41%), and heart failure with preserved ejection fraction (n = 12, 32%). There were seven patients with comorbid COPD; however, upon review of their chest CT scans, ILD was the predominant radiographic feature (i.e., these patients did not meet criteria for PH-COPD, and instead met criteria for CPFE as defined by the 2022 American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Asociacion Latinoamericana de Torax (ATS/ERS/JRS/ALAT) Research Statement [27]). The most frequent subtype of ILD was CTD-ILD (n = 27, 73%), followed by IPF (n = 4, 11%). Usual interstitial pneumonia (UIP) was the most common radiographic pattern (n = 17, 46%), followed by nonspecific interstitial pneumonia (NSIP) (n = 14, 38%).
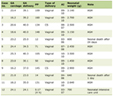 Click to view |
Table 1. Baseline Patient
Characteristics |
At baseline, prior to treatment with PAH-targeted therapy, the majority of patients had a WHO FC of III (n = 21, 68%), followed by FC II (n = 6, 19.4%). The mean 6MWD was 233 m (n = 10). PFTs were notable for a percent predicted TLC of 69±20%, percent predicted FVC of 60±16%, percent predicted forced expiratory volume in one second (FEV1) of 63±16% and percent predicted diffusion limitation of carbon monoxide (DLCO) of 35±15%. Baseline hemodynamic data were available in 33 patients and demonstrated a mPAP of 45 ± 11 mm Hg and PVR of 9 ± 4 WU. Baseline NT-proBNP was 1,498 ng/dL (n = 30, IQR 675 - 3,208).
The landscape of PAH-targeted therapy has evolved substantially during the course of this study resulting in inconsistencies in choice of PAH-targeted therapy in the patients. Majority of patients received treatment with a single PAH-targeted agent (n = 27, 73%), with PDE5-I being the most frequently utilized class in 51% of patients. Prostacyclins and ERAs were utilized less frequently (11% each). All nine patients on dual therapy received a combination of a PDE5-I and an ERA. Patients with most severe disease received therapy including infusion prostanoid (Table 2).
Click to view |
Table 2. Distribution of Pulmonary Arterial
Hypertension-Targeted Therapy |
The change in pulmonary hemodynamics, laboratory and functional parameters with PH-targeted therapy is summarized in Table 3. In patients with measurements both before and after treatment with PAH-targeted therapy, there was a reduction in NT-proBNP levels (n = 26, 1,421 vs. 842 ng/dL, P = 0.045) and PVR (n = 13, 8 vs. 5 WU, P < 0.001), and an increase in CO (n = 13, 4 vs. 5 L/min, P = 0.014) (Fig. 2). There was no significant difference in WHO FC, METs achieved on exercise stress test, PFT parameters, or 6MWD post-treatment (Fig. 3).
Click to view |
Table 3. Pulmonary Hemodynamics, Laboratory
Data, and Functional Parameters in Patients With Measurements Both Before and
After Treatment With Pulmonary Arterial Hypertension-Targeted
Therapy |
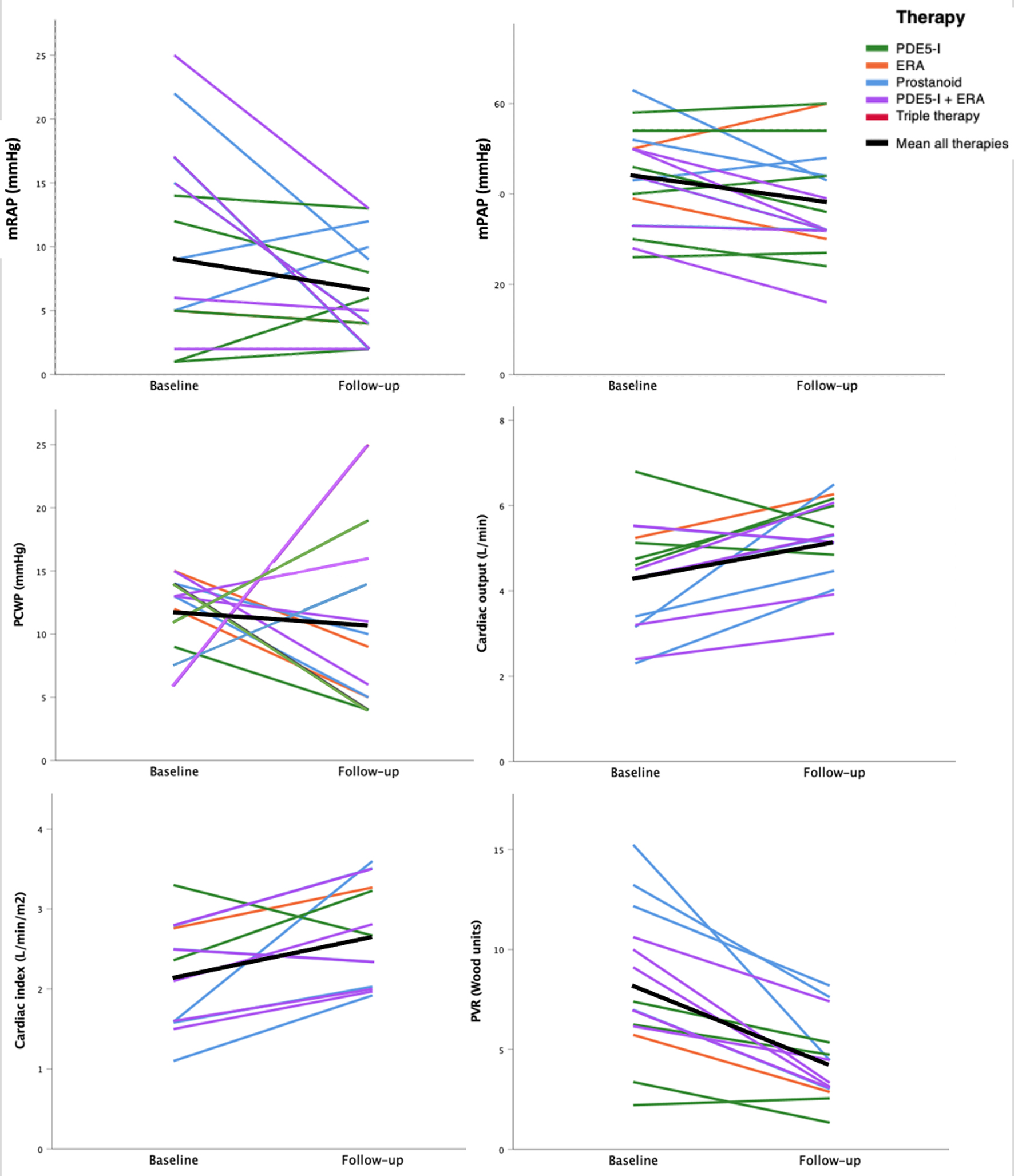 Click for large image |
Figure 2. Association between PAH-targeted therapy and hemodynamics. ERA: endothelin receptor antagonist; mPAP: mean pulmonary arterial pressure; mRAP: mean right atrial pressure; PCWP: pulmonary capillary wedge pressure; PDE5-I: phosphodiesterase-5 inhibitor; PVR: pulmonary vascular resistance. |
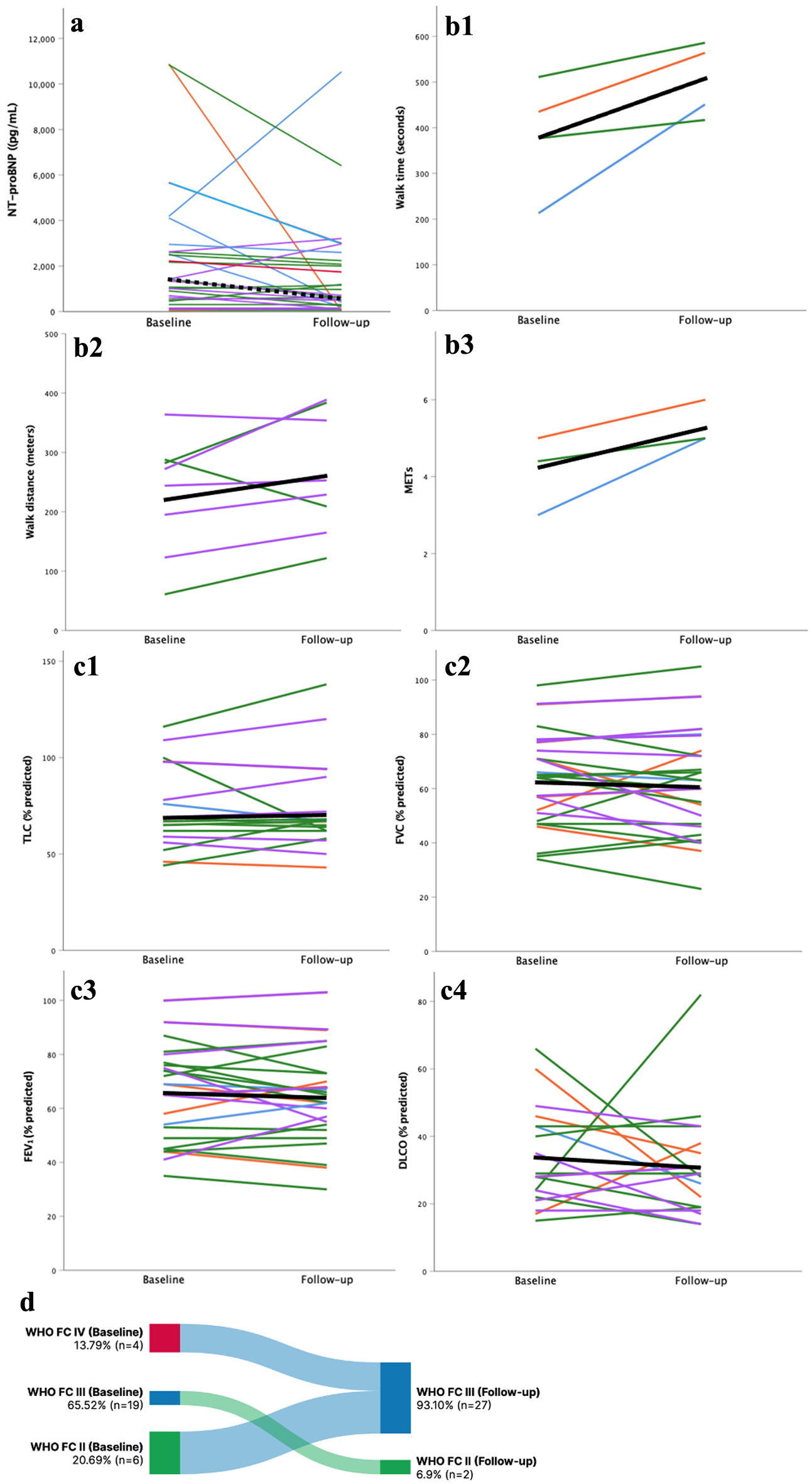 Click for large image |
Figure 3. Association between PAH-targeted therapy and NT-proBNP (a), exercise parameters (b1-3), PFTs (c1-4) and functional class (d). DLCO: diffusion limitation of carbon monoxide; ERA: endothelin receptor antagonist; FEV1: forced expiratory volume in one second; FVC: forced vital capacity; METs: metabolic equivalents of task; NT-proBNP: N-terminal pro-B-type natriuretic peptide; PDE5-I: phosphodiesterase-5 inhibitor; TLC: total lung capacity; WHO FC: World Health Organization functional class. |
Only one patient-reported side effect of therapy was captured (headache) which we believe is a flaw due to the retrospective nature of the study. Seventeen patients (46%) died with a median survival time of 5 years. Mortality was related to PH in 35% of patients (Supplementary Material 1, jocmr.elmerjournals.com).
| Discussion | ▴Top |
PAH-targeted therapy has been used in patients with PH-ILD despite limited evidence supporting their use. In this retrospective study, we demonstrate that the use of PAH-targeted therapy in a subset of patients with PH-ILD was associated with improved hemodynamic parameters. Of patients with available hemodynamic testing before and after treatment with PAH-targeted therapy, there was a significant reduction in PVR and mPAP and an increase in CO. Though RHC remains the gold standard for diagnosis of PH, hemodynamic data have been inconsistently reported across the available PH-ILD literature and were not evaluated in studies investigating the efficacy of bosentan, sildenafil, and macitentan [17-19, 22, 23]. A pilot trial published in 2012 showed that the use of riociguat led to improvement in PVR and CO, but these findings did not reach statistical significance and the subsequent RISE-IIP randomized controlled trial (RCT) revealed significant harm and increased mortality associated with use in PH-ILD [20, 21]. While our study was underpowered to detect associations between individual classes of PAH-targeted therapies and hemodynamics, we hope the significant hemodynamic improvement noted in this cohort will stimulate further discussion that may lead to further investigation with prospective RCTs.
We also noted a significant reduction in NT-proBNP after treatment with PAH-targeted therapy which persisted even after exclusion of the two outliers (Fig. 3). Several studies have demonstrated that plasma NT-proBNP levels are an independent predictor of survival in PH and correlate with functional capacity, right ventricular function, and hemodynamic variables, and the latter often times may not be available within the ILD population due to the risk of ILD exacerbation associated with procedural interventions [16]. In their 2021 RCT evaluating inhaled treprostinil in patients with ILD, Waxman and colleagues also reported a decrease in NT-proBNP levels by 15% in the treatment group, compared to a 46% increase in the placebo group (treatment ratio 0.58, 95% confidence interval 0.47 - 0.72, P < 0.001) [15]. Similarly, in a small study of 15 patients with PH-ILD, Corte et al found a significant reduction in BNP levels (P = 0.03) after 6 months of therapy with sildenafil [24]. Despite Corte and colleagues’ promising results, subsequent RCTs investigating the use of sildenafil in PH-ILD did not report NT-proBNP levels, and of the three published studies investigating the use of bosentan in IPF, including one large RCT, none collected information on NT-proBNP levels at baseline or following treatment [17-19, 22].
Functional response to PAH-targeted therapy is reported consistently across the literature. The 6MWD is frequently used as the primary end-point of studies within the PH literature as walk tests are well-tolerated, inexpensive, and can be repeated easily in the clinic to assess response to treatment. Our patients did not have a significant increase in 6MWD following PAH-targeted therapy, though only a small proportion of patients had available 6MWD data. Small studies have demonstrated improvement in 6MWD with the use of sildenafil; however, the larger STEP-IPF RCT did not meet their primary outcome of a 20% improvement in 6MWD [22, 24, 26]. They did, however, find a significant improvement in dyspnea score and on quality of life questionnaire in the treatment group [22]. In keeping with our findings, neither riociguat or bosentan have been shown to improve 6MWD, dyspnea scores, or quality of life scores [17, 18, 20]. Recently, inhaled treprostinil was found to increase 6MWD by 31 m (95% confidence interval 16.85 - 45.39, P < 0.001), a finding that has led to increasing use of this medication in patients with PH-ILD [15]. Typically, patients with ILD experience a progressive decline in functional status even in the absence of PH. We postulate that the relative stability in walk distance throughout follow-up may in fact represent a benefit of PAH-targeted therapy in our patients.
As clinicians consider the use of PAH-targeted therapy in patients with PH-ILD, it is important to determine whether these medications are safe and tolerable. As our study was retrospective in nature, we were unable to collect information regarding adverse events but did note that only one patient in our cohort reported a side effect of treatment, which we suspect was likely a flaw of incomplete documentation in the electronic medical record. Several studies have demonstrated that most classes of PAH-targeted therapies are relatively well tolerated and have limited adverse events, with the notable exception of ERAs and riociguat, which carry a Blackbox warning for embryo-fetal toxicity [15, 17-21, 28, 29]. Equally concerning are the increased rates of death in the treatment group reported by Nathan and colleagues that ultimately led to the early termination of their 2019 RCT and a strong recommendation against use of riociguat in patients with PH-ILD [21]. Notably, our study was a retrospective cohort study and no patients were initiated on ERAs or riociguat after the Blackbox warning was issued.
This exploratory study had several limitations. First, the sample size was small and our study was underpowered to perform subgroup analysis of ILD subtype and/or PAH-targeted therapy class. However, PAH-targeted therapy is not used widely in patients with PH-ILD due to the lack of current FDA approval for this indication, and there are limited studies investigating use in this patient population [30, 31]. Due to the retrospective nature of this study, data collection was limited by lack of available data both before and after treatment with PAH-targeted therapy, including laboratory values functional parameters, and hemodynamic data. While this heterogeneity may have led to effect modification and individual treatment bias, our cohort was ultimately reflective of a real-world experience in which diagnostics may not be obtained in a prescribed schedule. Additionally, the 2018 WSPH definition for pre-capillary PH was used in this study (PVR > 3 WU), which has since been updated by the European Society of Cardiology/European Respiratory Society (ESC/ERS) guidelines as a PVR > 2 WU [1, 32]. At present, there is a scarcity of data evaluating the efficacy of PAH therapy in patients with a PVR between 2 and 3 WU; as such, we believe using a cutoff of PVR > 3 WU was more appropriate. Finally, accurate reporting and capture of side effects was likely limited.
Conclusion
The use of PAH-targeted therapy may be associated with improved hemodynamics and a reduction in NT-proBNP levels in a subset of patients with PH-ILD, and the results of this study raise clinical equipoise for further study. The future application of precision medicine to aid in phenotyping patients with WSPH group 3 PH may help clinicians better understand which patients are likely to benefit from PAH-targeted therapy [33]. Additionally, given what is known about the beneficial effects of inhaled treprostinil in patients with PH-ILD, studies evaluating the route of administration of PAH-targeted therapy is also warranted.
| Supplementary Material | ▴Top |
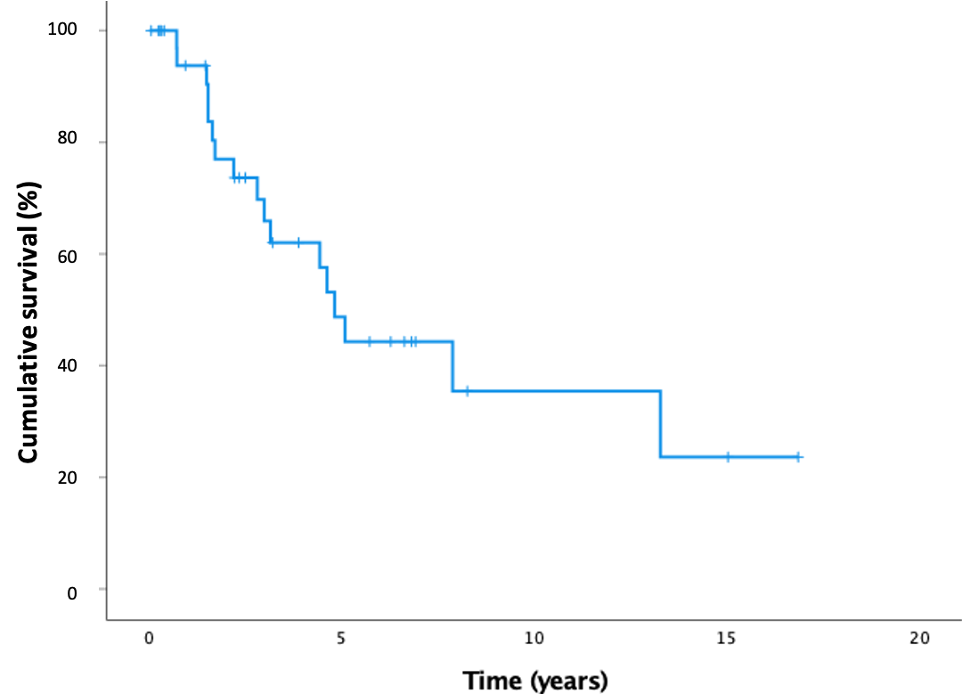
Suppl 1. Kaplan-Meier survival curve. With overall survival defined as time to death or transplant. Median survival time was 4.81 years.
{kind=link}
Acknowledgments
None to declare.
Financial Disclosure
Kavitha Selvan was supported by the NIH/NHLBI Institutional National Research Service Award T32 HL007605.
Conflict of Interest
Remzi Bag received research grants from United Therapeutics, Medtronic, Reata Pharmaceuticals, Gilead, Liquidia, PhaseBio, Actelion, and Bayer and received speaker and consultant honoraria from Bayer. Kavitha Selvan and Krittika Teerapuncharoen have no conflict of interest to disclose.
Informed Consent
Written informed consent was obtained from all participants for inclusion in the research registry. The institutional review board determined that no additional informed consent was required for this study.
Author Contributions
Remzi Bag is the guarantor of this manuscript. Kavitha Selvan, Krittika Teerapuncharoen, and Remzi Bag contributed substantially to the study design, interpretation of the data, and review and revision of the manuscript, and had full access to all of the data in this study and take responsibility for the integrity of the data. Krittika Teerapuncharoen was responsible for data analysis and takes responsibility for the accuracy of analysis. Kavitha Selvan contributed substantially to the writing of the manuscript.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
Abbreviations
6MWD: six-minute walk distance; ACEIs/ARBs: angiotensin-converting enzyme inhibitors/angiotensin receptor blockers; ATS/ERS/JRS/ALAT: American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Asociacion Latinoamericana de Torax; CHD: congenital heart disease; CHF: congestive heart failure; CI: cardiac index; CO: cardiac output; COPD: chronic obstructive pulmonary disease; CPFE: combined pulmonary fibrosis and emphysema; CT: computed tomography; CTD: connective tissue disease; CTD-ILD: connective tissue disease-associated ILD; CTEPH: chronic thromboembolic pulmonary hypertension; DLCO: diffusion limitation of carbon monoxide; ERA: endothelin receptor antagonist; FC: functional class; FDA: Food and Drug Administration; FEV1: forced expiratory volume in one second; FVC: forced vital capacity; HRCT: high-resolution computed tomography; IPF: idiopathic pulmonary fibrosis; ILA: interstitial lung abnormality; ILD: interstitial lung disease; IQR: interquartile range; IV: intravenous; METs: metabolic equivalents of task; mPAP: mean pulmonary artery pressure; mRAP: mean right atrial pressure; NSIP: nonspecific interstitial pneumonia; NT-proBNP: N-terminal pro-B-type natriuretic peptide; PCWP: pulmonary capillary wedge pressure; PDE5-I: phosphodiesterase-5 inhibitor; PFT: pulmonary function test; PH: pulmonary hypertension; PH-ILD: pulmonary hypertension due to interstitial lung disease; PLCH: pulmonary Langerhans cell histiocytosis; PoPH: portopulmonary hypertension; PVR: pulmonary vascular resistance; RCT: randomized controlled trial; RHC: right heart catheterization; SD: standard deviation; sGCS: soluble guanylyl cyclase stimulator; SLE: systemic lupus erythematosus; TLC: total lung capacity; UIP: usual interstitial pneumonia; WHO: World Health Organization; WSPH: The World Symposium on Pulmonary Hypertension; WU: Wood units
| References | ▴Top |
- Condon DF, Nickel NP, Anderson R, Mirza S, de Jesus Perez
VA. The 6th World Symposium on Pulmonary Hypertension: what's old is new. F1000Res. 2019;8(F1000
Faculty Rev):888.
doi pubmed - Behr J, Ryu JH. Pulmonary hypertension in interstitial lung
disease. Eur Respir J. 2008;31(6):1357-1367.
doi pubmed - Ruffenach G, Hong J, Vaillancourt M, Medzikovic L, Eghbali
M. Pulmonary hypertension secondary to pulmonary fibrosis: clinical data, histopathology and
molecular insights. Respir Res. 2020;21(1):303.
doi pubmed - Almaaitah S, Highland KB, Tonelli AR. Management of
pulmonary arterial hypertension in patients with systemic sclerosis. Integr Blood Press Control.
2020;13:15-29.
doi pubmed - Fartoukh M, Humbert M, Capron F, Maitre S, Parent F, Le Gall
C, Sitbon O, et al. Severe pulmonary hypertension in histiocytosis X. Am J Respir Crit Care Med.
2000;161(1):216-223.
doi pubmed - Nadrous HF, Pellikka PA, Krowka MJ, Swanson KL, Chaowalit N,
Decker PA, Ryu JH. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest.
2005;128(4):2393-2399.
doi pubmed - Andersen CU, Mellemkjaer S, Hilberg O, Nielsen-Kudsk JE,
Simonsen U, Bendstrup E. Pulmonary hypertension in interstitial lung disease: prevalence,
prognosis and 6 min walk test. Respir Med. 2012;106(6):875-882.
doi pubmed - Lv H, Liu J, Pan Q, Cai R, Zhang J. Clinical retrospective
analysis of interstitial lung disease patients associated with pulmonary hypertension. Med Sci
Monit. 2019;25:7763-7769.
doi pubmed - Hervier B, Meyer A, Dieval C, Uzunhan Y, Devilliers H, Launay
D, Canuet M, et al. Pulmonary hypertension in antisynthetase syndrome: prevalence, aetiology and
survival. Eur Respir J. 2013;42(5):1271-1282.
doi pubmed - Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF.
Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary
fibrosis. Chest. 2006;129(3):746-752.
doi pubmed - Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD.
Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur
Respir J. 2007;30(4):715-721.
doi pubmed - Nathan SD, Cottin V. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Eur Respir Monogr. 2012;57:148-160.
- Cottin V. Treatment of pulmonary hypertension in
interstitial lung disease: do not throw out the baby with the bath water. Eur Respir J.
2013;41(4):781-783.
doi pubmed - Alhamad EH, Cal JG, Alrajhi NN, Alharbi WM. Predictors of
mortality in patients with interstitial lung disease-associated pulmonary hypertension. J Clin
Med. 2020;9(12):3828.
doi pubmed - Waxman A, Restrepo-Jaramillo R, Thenappan T, Ravichandran A,
Engel P, Bajwa A, Allen R, et al. Inhaled treprostinil in pulmonary hypertension due to
interstitial lung disease. N Engl J Med. 2021;384(4):325-334.
doi pubmed - Chin KM, Rubin LJ, Channick R, Di Scala L, Gaine S, Galie N,
Ghofrani HA, et al. Association of N-terminal pro brain natriuretic peptide and long-term
outcome in patients with pulmonary arterial hypertension. Circulation.
2019;139(21):2440-2450.
doi pubmed - King TE, Jr., Behr J, Brown KK, du Bois RM, Lancaster L, de
Andrade JA, Stahler G, et al. BUILD-1: a randomized placebo-controlled trial of bosentan in
idiopathic pulmonary fibrosis. Am J Respir Crit Care Med.
2008;177(1):75-81.
doi pubmed - King TE, Jr., Brown KK, Raghu G, du Bois RM, Lynch DA,
Martinez F, Valeyre D, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic
pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(1):92-99.
doi pubmed - Gunther A, Enke B, Markart P, Hammerl P, Morr H, Behr J,
Stahler G, et al. Safety and tolerability of bosentan in idiopathic pulmonary fibrosis: an open
label study. Eur Respir J. 2007;29(4):713-719.
doi pubmed - Hoeper MM, Halank M, Wilkens H, Gunther A, Weimann G, Gebert
I, Leuchte HH, et al. Riociguat for interstitial lung disease and pulmonary hypertension: a
pilot trial. Eur Respir J. 2013;41(4):853-860.
doi pubmed - Nathan SD, Behr J, Collard HR, Cottin V, Hoeper MM, Martinez
FJ, Corte TJ, et al. Riociguat for idiopathic interstitial pneumonia-associated pulmonary
hypertension (RISE-IIP): a randomised, placebo-controlled phase 2b study. Lancet Respir Med.
2019;7(9):780-790.
doi pubmed - Idiopathic Pulmonary Fibrosis Clinical Research Network,
Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, Hunninghake GW. A controlled trial of
sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med.
2010;363(7):620-628.
doi pubmed - Raghu G, Million-Rousseau R, Morganti A, Perchenet L, Behr
J, MUSIC Study Group. Macitentan for the treatment of idiopathic pulmonary fibrosis: the
randomised controlled MUSIC trial. Eur Respir J. 2013;42(6):1622-1632.
doi pubmed - Corte TJ, Gatzoulis MA, Parfitt L, Harries C, Wells AU, Wort
SJ. The use of sildenafil to treat pulmonary hypertension associated with interstitial lung
disease. Respirology. 2010;15(8):1226-1232.
doi pubmed - Behr J, Nathan SD, Wuyts WA, Mogulkoc Bishop N, Bouros DE,
Antoniou K, Guiot J, et al. Efficacy and safety of sildenafil added to pirfenidone in patients
with advanced idiopathic pulmonary fibrosis and risk of pulmonary hypertension: a double-blind,
randomised, placebo-controlled, phase 2b trial. Lancet Respir Med. 2021;9(1):85-95.
doi pubmed - Collard HR, Anstrom KJ, Schwarz MI, Zisman DA. Sildenafil
improves walk distance in idiopathic pulmonary fibrosis. Chest. 2007;131(3):897-899.
doi pubmed - Cottin V, Selman M, Inoue Y, Wong AW, Corte TJ, Flaherty KR,
Han MK, et al. Syndrome of combined pulmonary fibrosis and emphysema: an official
ATS/ERS/JRS/ALAT research statement. Am J Respir Crit Care Med.
2022;206(4):e7-e41.
doi pubmed - Opsumit. 2022. Opsumithcp.com.
- Adempas. 2022. Adempashcp.com.
- Dawes TJW, McCabe C, Dimopoulos K, Stewart I, Bax S, Harries
C, Samaranayake CB, et al. Phosphodiesterase 5 inhibitor treatment and survival in interstitial
lung disease pulmonary hypertension: A Bayesian retrospective observational cohort study.
Respirology. 2023;28(3):262-272.
doi pubmed - Chauvelot L, Gamondes D, Berthiller J, Nieves A, Renard S,
Catella-Chatron J, Ahmad K, et al. Hemodynamic response to treatment and outcomes in pulmonary
hypertension associated with interstitial lung disease versus pulmonary arterial hypertension in
systemic sclerosis: data from a study identifying prognostic factors in pulmonary hypertension
associated with interstitial lung disease. Arthritis Rheumatol. 2021;73(2):295-304.
doi pubmed - Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF,
Brida M, Carlsen J, et al. [2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary
hypertension]. G Ital Cardiol (Rome). 2023;24(4 Suppl 1):e1-e116.
doi pubmed - Singh N, Dorfmuller P, Shlobin OA, Ventetuolo CE. Group 3
pulmonary hypertension: from bench to bedside. Circ Res. 2022;130(9):1404-1422.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Clinical Medicine Research is published by Elmer Press Inc.