| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website https://jocmr.elmerjournals.com |
Original Article
Volume 17, Number 4, April 2025, pages 231-239

Phenotypic Variability and Hematological Characterization of β0- and β+-Thalassemia Carriers: A Comparative Study
Ahmad Al Tibia, Diya Hasanb, h ![]() , Ola M. Al-Sanabrac, Ghaith H. Mansourb, Maissa T. Shawagfehb, Moath Alqaralehc, Tareq Nayef AlRamadnehd, Mutaz Jamal Al-Khreisate, George J. Burghelf, g, Amid Abdelnoura
, Ola M. Al-Sanabrac, Ghaith H. Mansourb, Maissa T. Shawagfehb, Moath Alqaralehc, Tareq Nayef AlRamadnehd, Mutaz Jamal Al-Khreisate, George J. Burghelf, g, Amid Abdelnoura
aBiolab Diagnostic Laboratories, Amman, Jordan
bDepartment of Allied Medical Sciences, Zarqa College, Al-Balqa Applied University, Zarqa, Jordan
cDepartment of Medical Laboratory Sciences, Faculty of Allied Medical Sciences, Al-Balqa Applied University, Al-Salt 19117, Jordan
dDepartment of Medical Laboratory Sciences, Faculty of Allied Medical Sciences, Zarqa University, Al-Zarqa 13132, Jordan
eDepartment of Medical Laboratory Sciences, Faculty of Allied Medical Sciences, Al-Ahliyya University, Amman 19328, Jordan
fManchester Centre for Genomic Medicine and NW Laboratory Genetics Hub, Manchester University Hospitals NHS Foundation Trust, Manchester, UK
gDivision of Cancer Sciences, School of Medical Sciences, Faculty of Biology, Medicine and Health, The University of Manchester, Manchester, UK
hCorresponding Author: Diya Hasan, Department of Allied Medical Sciences, Zarqa College, Al-Balqa Applied University, Zarqa, Jordan
Manuscript submitted February 21, 2025, accepted April 10, 2025, published online April 19, 2025
Short title: β-Thalassemia Variants
doi: https://doi.org/10.14740/jocmr6213
| Abstract | ▴Top |
Background: β-Thalassemia is a genetic disorder characterized by decreased or completely absent β-globin synthesis, leading to a spectrum of clinical manifestations. It is a major public health concern in Jordan, as in other Mediterranean countries. β-Thalassemia carriers are normally asymptomatic; nevertheless, laboratory examinations often reveal mild anemia characterized by microcytic hypochromic erythrocytes, with differences influenced by specific phenotypes. This study aimed to assess and correlate the variants among β0 and β+ phenotypes in the Jordanian population with hematological characteristics, as well as establish and determine reference values for distinguishing between the two phenotypes.
Methods: One hundred forty-five β-thalassemia carriers were recruited from various governorates in Jordan. Hematological parameters, including complete blood count (CBC) and capillary electrophoresis of hemoglobin (Hb), were evaluated in all participants. Molecular techniques, specifically polymerase chain reaction (PCR) with hybridization, were employed to identify β-thalassemia variants and classify the participants as having β0 and β+ phenotypes.
Results: Among the 145 β-thalassemia carriers, 64 (44.14%) and 81 (55.86%) had β0-thalassemia and β+-thalassemia, respectively. Participants exhibiting a cutoff value of Hb (≤ 11.0 g/dL), mean corpuscular volume (MCV) (≤ 64.0 fL), mean corpuscular hemoglobin (MCH) (≤ 19.0 pg), and hemoglobin A2 (Hb-A2) (≥ 5.00%) were classified as having the β0 phenotype. These participants demonstrated significantly lower mean Hb, MCV, MCH, and higher mean Hb-A2 than the participants with the β+ phenotype (P < 0.0001).
Conclusions: Hb, MCV, MCH, and Hb-A2 can serve as effective screening tools for predicting β0- and β+-thalassemia in the Jordanian population. These findings have important clinical implications for early diagnosis, genetic counseling, and prenatal screening of β-thalassemia.
Keywords: Hemoglobinopathies; β-thalassemia; Pathogenic variants; Phenotype; Hemoglobin variants
| Introduction | ▴Top |
Jordan, a Middle Eastern country bordering Saudi Arabia, Iraq, Syria, and Palestine, has a population of approximately 11 million, primarily concentrated in urban areas, and consists of 12 governorates; Amman houses around 40% of the population, while Irbid (an academic and agricultural hub) and Zarqa (an industrial center) are also major population centers. Mafraq has grown rapidly due to the refugee influx, while Balqa, Karak, and Maan hold historical significance [1].
One of the most prevalent monogenic illnesses worldwide is thalassemia [2], which mostly affects people in the Mediterranean region, Southeast Asia, and the Middle East, including Jordan, Palestine, Syria, Lebanon, Egypt, and Iraq. Reduced hemoglobin (Hb) production is a hallmark of this genetic condition and can result in anemia and other problems [3]. Owing to its frequency and clinical importance, β-thalassemia is distinguished from the other forms; it results from variants in the HBB gene, which codes for the β-globin component that disrupts globin chain synthesis, leading to β0-thalassemia (β0) resulting in a complete absence of β-globin production, or β+-thalassemia (β+) resulting in a partial reduction in β-globin synthesis [4]. β-Thalassemia occurs in various severities, from mild variants that require little treatment to severe versions that require frequent blood transfusions and continuous medical attention. To effectively manage and cure β-thalassemia and enhance the quality of life for those who have it, comprehending its genetic and clinical components is essential [5]. In 2021, the World Health Organization (WHO) reported that the carrier rate for β-thalassemia in the Middle East ranged from 3% to 10%, with 1.1% of couples at risk of passing the condition to their children [6, 7].
Depending on the age at diagnosis and disease severity, β-thalassemia has a wide range of clinical variances. Although thalassemia carriers are asymptomatic, laboratory tests can reveal variations in Hb levels and microcytic hypochromic erythrocytes [8]. Moreover, analysis for the Hb will show elevated hemoglobin F (Hb-F) and hemoglobin A2 (Hb-A2) level, but in some cases, the Hb-F level can be normal. In 2004, Jordan started a mandatory National Premarital Thalassemia Screening Program to detect thalassemia carriers, inform them of the risks, and help prevent new cases. Mean corpuscular volume (MCV) should be regularly used as a mandatory screening tool before marriage [6].
According to these regulations, people with < 80 fL MCV and < 13 g/dL Hb should undergo additional evaluation [9]. The Hb-A2 test is used to determine whether they are thalassemia carriers: a level between 3.6% and 4.2% indicates mild β+-thalassemia, while a level between 4% and 9% indicates heterozygote β0-thalassemia and severe β+-thalassemia [10].
Over 200 distinct pathogenic variations impact varying amounts of β-globin gene expression [11]. These genetic variations are not evenly distributed among individuals and follow unique racial and regional patterns [12]. Each population is distinguished by a few common mutations and various rare variants, reflecting the genetic diversity shaped by historical and geographic influences [13]. Insertions or deletions in HBB can result in nucleotide substitutions or frameshift mutations that affect β-globin mRNA transcription, splicing, or translation [14].
According to current reports, thalassemia carrier incidence in Jordan is approximately 2-4%. Six hundred fifty-six patients (550 with major β-thalassemia and 106 with intermedia β-thalassemia) are recorded cases medicated by the Health Ministry [7]. According to the national registry, 1,228 patients were registered in the centers of thalassemia remedies under the Health Ministry supervision in 2017, representing approximately 86% of the 1,450 patients with thalassemia in Jordan [6, 10]. Therefore, based on the β-thalassemia phenotypes β0 and β+, this work primarily aimed to create suitable reference values for hematological parameters and Hb levels and investigate any notable variations in hematological traits between these two phenotypes.
| Materials and Methods | ▴Top |
Ethical approval and participant cohort eligibility
This study was approved by the institutional review board (IRB) of Al-Balqa Applied University, Jordan (26/3/2/274). The study was conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration.
Blood samples were screened for MCV. The samples with MCV < 80 fL underwent Mentzer Index calculation (MCV/red blood cell (RBC) count), where a Mentzer Index < 13 indicated likely β-thalassemia carrier, while > 13 suggested iron deficiency anemia (IDA). Patients with normal Hb levels and MCV were excluded from this study.
Whole blood from 145 thalassemia carriers from different Jordanian governments was collected and comprehensively examined, including capillary electrophoresis of Hb and complete blood count (CBC). Furthermore, a genetic investigation was performed to determine β-thalassemia variants and phenotypes. The geographical distribution data for the β-thalassemia carriers were collected from 10 governorates representing various parts of Jordan. The samples were obtained from people who visited the Biolab Medical Laboratory and samples referred from other laboratories in Jordan.
Fifty samples were also collected from healthy individuals with no risk factors that could affect blood indices, such as heart disease, kidney or liver disease, or medication use, as confirmed through a questionnaire.
Hematological measurements
Whole blood samples (4 mL) were collected from patients in ethylenediaminetetraacetic acid (EDTA) tubes for routine hematological examinations, including capillary electrophoresis of Hb using MINICAP (Sebia®, Lisses, France) and CBC using Sysmex XN-1000 (Sysmex Corporation®, Kobe, Japan).
DNA extraction and multiplex polymerase chain reaction (PCR)
DNA from the blood samples was extracted by automated EZ1 instrument (Qiagen®, Hilden, Germany) and quantified using the Qubit 4 fluorometer and dsDNA high-sensitivity test kit (Thermo Fisher Scientific®, USA) for DNA concentration measurement in ng/µL. All protocols were performed according to the manufacturer’s instructions.
DNA was amplified using the multiplex PCR (Veriti Thermal 96-Well Thermal Cycler Applied Biosystems®, USA) system, and a reverse hybridization-based commercial β-globin test strip was used to detect the 22 β-globin gene targets (Vienna Lab Labor diagnostika GmbH®, Austria). The StripAssay® Online Calculator v2.17 was utilized to analyze the results. The samples were processed at Biolab Diagnostic Laboratories (Amman, Jordan).
Statistical analysis
GraphPad Prism and Microsoft Excel were used for data analysis. The raw data were recorded and arranged using an Excel spreadsheet and then imported by GraphPad Prism for statistical analysis. Data were presented as either mean ± standard deviation (SD) or median. t-test and one-way analysis of variance (ANOVA) were performed for comparing two or more groups, respectively. The data are considered significant if the P-value is less than or equal to 0.05.
| Results | ▴Top |
This study consists of 145 thalassemia carriers. CBC and capillary electrophoresis of Hb data were collected from all participants. Table 1 shows a list of participants’ characteristics. Males made up 47.6%, and females made up 52.4%. Participants’ ages ranged from 1 to 80 years, with a median age of 21 years.
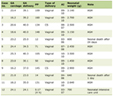 Click to view | Table 1. Patient Gender and Age Distribution With Hematological Parameters Among β0- and β+-Thalassemia Phenotypes and Control Groups |
The population distribution of the study group in Jordanian governorates was as follows: Amman 62.75%, Irbid 15.86%, Zarqa 9.66%, Maan 2.76%, Tafilah and Mafraq 2.07% each, Madaba, Aqaba, and Jarash 1.38% each, and Balqa 0.69% (Table 2). Sixteen β-thalassemia variants were tested in the study population; 64 participants with β0 phenotype (44.14%) and 81 participants with β+ phenotype (55.86%) were heterozygous variant carriers, and all variants were classified as pathogenic. In this study, no compound heterozygous or homozygous variants were detected. The distribution and percentage were calculated for each variant based on the phenotype classification. β0 phenotypes included IVS 2.1 [G>A] (32.8%), IVS 1.1 [G>A] (28.1%), codon 8 [-AA] (12.5%), codon 5 [-CT] (10.9%), and codon 39 [C>T] (10.9%) each; codon 6 [-A], IVS 1.130 [G>C], and codon 44 [-C] (1.6%) each. β+ phenotypes included IVS 1.6 [T>C] (34.6%), IVS 1.110 [G>A] (25.9%), IVS 2.745 [C>G] (19.8%), IVS 1.5 [G>C] (6.2%), -30 [T>A] (4.9%), -87 [C>G] (3.7%), -101 [C>T] (3.7%), and IVS 2.848 [C>A] (1.2%) (Table 3).
Click to view | Table 2. Population Distribution in Jordan’s Governorates |
Click to view | Table 3. β-Thalassemia Variants and Phenotypes (β0 and β+) Distribution |
The Hb analysis (Hb-A2) and hematological parameter (Hb and MCV) distribution for each phenotype in our study were shown among three groups: patients with β0-thalassemia (N = 64), patients with β+-thalassemia (N = 81), and a healthy participant group (N = 50). The results revealed that mean Hb levels vary significantly between study groups: β0-thalassemia 11.2 ± 0.57 g/dL, β+-thalassemia 12.5 ± 0.62 g/dL, and control group 14.44 ± 1.29 g/dL, with cutoff ≤ 11.0 g/dL to consider a β0-thalassemia case (P < 0.0001); the MCV was significantly different in study groups: 64.4 ± 2.8 fL in β0-thalassemia, 70.3 ± 2.4 fL in β+-thalassemia, and 85.96 ± 3.30 fL in the control group, with cutoff ≤ 64.0 fL to consider a β0-thalassemia case (P < 0.0001). Also, the MCH was significantly different in groups: 18.9 ± 1.6 pg in β0-thalassemia, 22.8 ± 2.6 fL in β+-thalassemia, and 32.3 ± 2.57 fL in the control group, with a cutoff ≤ 19.0 pg to consider a β0-thalassemia case (P < 0.0001). Despite these, the Hb-A percentage for β0-thalassemia, β+-thalassemia, and healthy participants was 93.5±0.96%, 89.5±5.5%, and 97.55±0.35%, respectively (P = 0.074), indicating no significant difference. However, the Hb-A2 was significantly upregulated in thalassemia (5.06±0.33%, 4.2±0.32%, and 2.41±0.378% for β0-thalassemia, β+-thalassemia, and healthy participants, respectively), with ≥ 5.00% cutoff to consider a β0-thalassemia case (P < 0.0001). Finally, the Hb-F percentages were as follows: 2.1±1.9% for β0-thalassemia, 1.8±1.6% for β+-thalassemia, and 0.98±0.89% for healthy participants (P = 0.050), which is near narrated significant. In conclusion, these results indicated that the hematological differences between the phenotypes of various types of thalassemia patients and healthy individuals are significant (Table 1), and in detail for each variant as shown in Table 4.
Click to view | Table 4. Distribution of Hematological Parameters and Hb Analysis Among β0- and β++-Thalassemia Phenotypes |
Figure 1 displays statistically significant variances in the mean, Hb, MCV, and MCH. Figure 2 shows the significance for Hb-A2. Hb, MCV, and MCH levels were lower, and Hb-A2 was higher in β0-thalassemia phenotypes in comparison to β+-thalassemia phenotypes, with P < 0.0001.
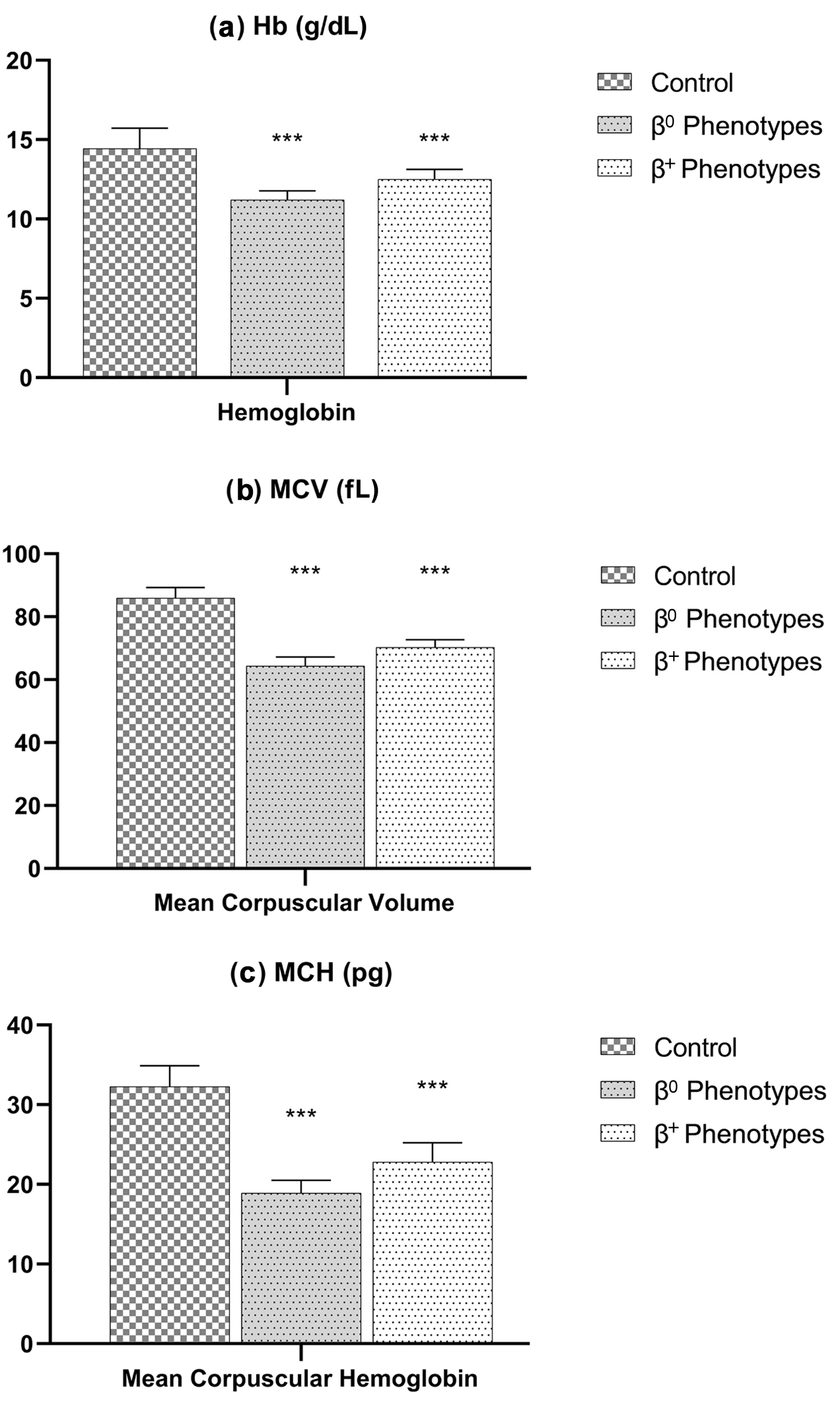 Click for large image | Figure 1. Hematological parameters (a: Hb; b: MCV; c: MCH) among β0- and β+-thalassemia phenotypes. Both thalassemic phenotypes have a significant (P-value < 0.0001) lower hemoglobin, MCV, and MCH compared to the control group. Hb: hemoglobin; MCH: mean corpuscular hemoglobin; MCV: mean corpuscular volume. |
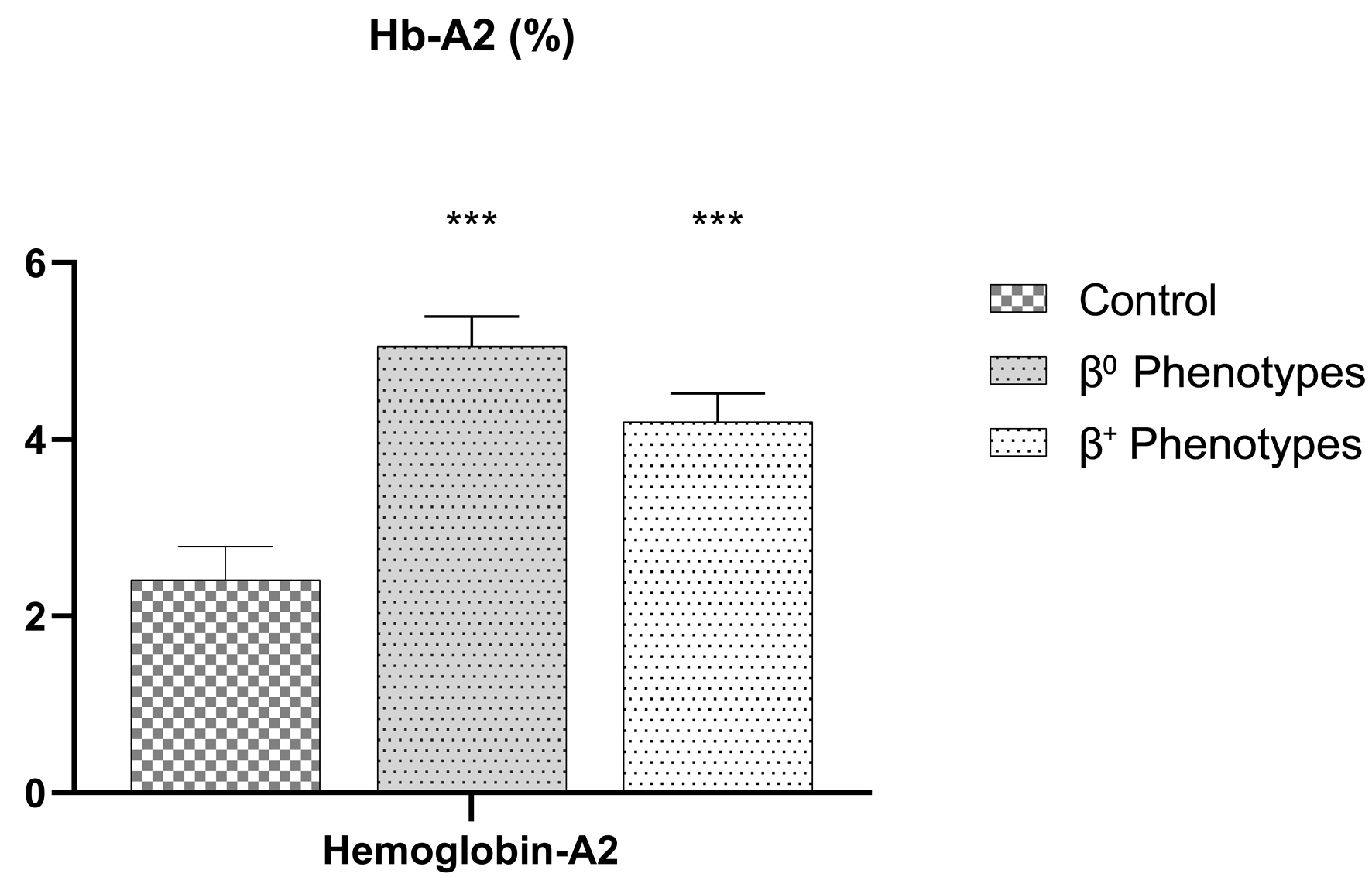 Click for large image | Figure 2. Capillary electrophoresis of Hb-A2 level among β0- and β+-thalassemia phenotypes. Both thalassemic phenotypes have a significant (P-value < 0.0001) higher Hb-A2 compared to the control group. Hb-A2: hemoglobin A2. |
Figure 3 presents a dot plot comparing Hb, MCV, MCH, and Hb-A2 levels between the two phenotypes, β0-thalassemia and β+-thalassemia, as well as the control group.
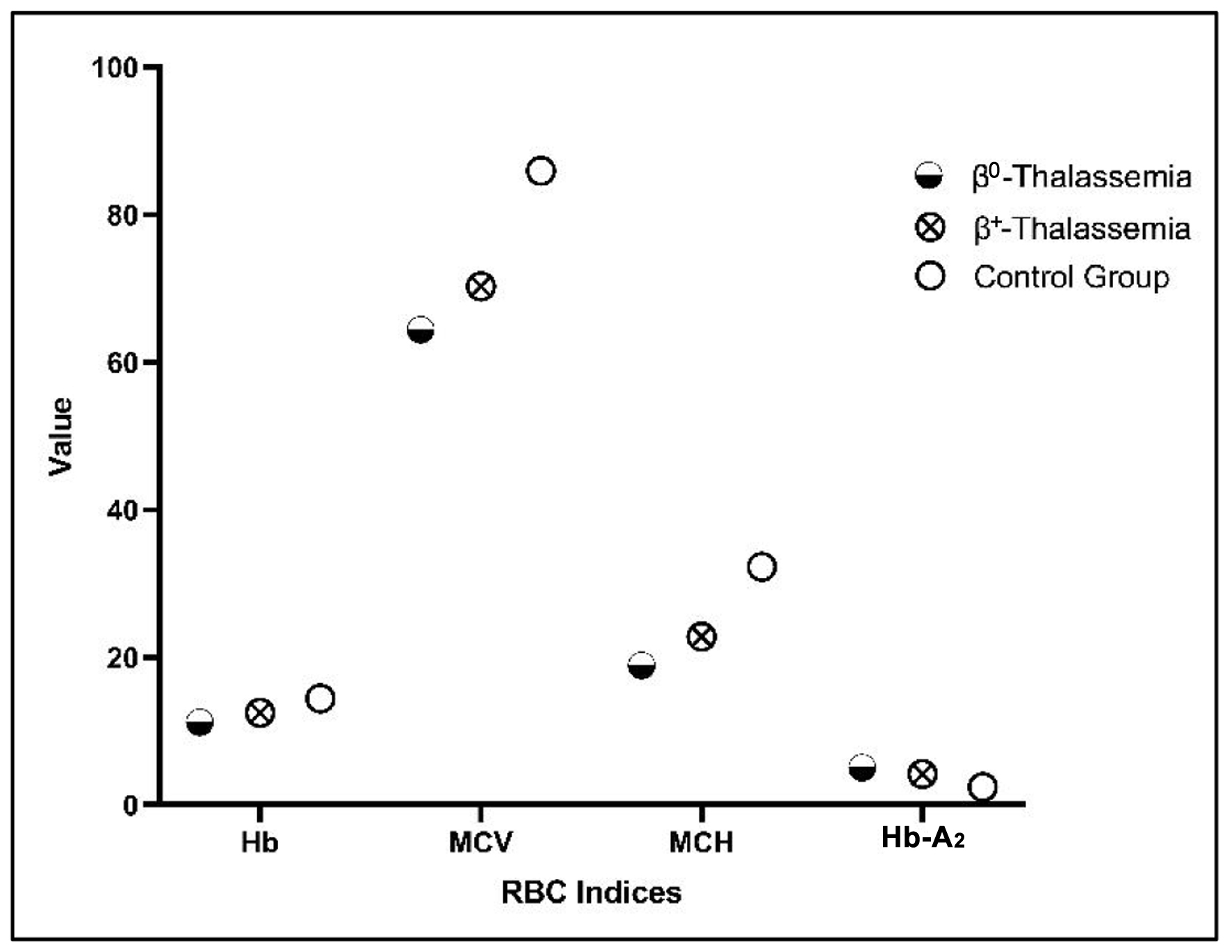 Click for large image | Figure 3. Dot plot comparing Hb, MCV, MCH, and Hb-A2 levels among β0- and β+-thalassemia phenotypes. The dot plot visualizes the differences between the β0-thalassemia and β+-thalassemia phenotypes compared to the control group. Hb: hemoglobin; Hb-A2: hemoglobin A2; MCH: mean corpuscular hemoglobin; MCV: mean corpuscular volume. |
These data may help determine the cutoff for each variable by clarifying the distinctions between the two phenotypes. The β0 phenotype was identified in patients with Hb cutoff values of ≤ 11.0 g/dL, MCV of ≤ 64.0 fL, MCH ≤ 19.0 pg, and Hb-A2 of ≥ 5.00%. Compared to the patients with the β+ phenotype, these patients showed considerably lower mean Hb, MCV, and MCH levels and higher mean Hb-A2 (P < 0.0001).
| Discussion | ▴Top |
Thalassemia is a serious health problem worldwide, with predominant prevalence in the Middle East, Southeast Asia, and Mediterranean [13]. This is mainly due to interethnic and consanguinity marriages, which contribute to variant pooling and cause this disorder in these regions of the world [3]. Thus, effective screening is a crucial approach to avoid the birth of infants with thalassemia major in Jordan, considering that the β-thalassemia carrier rate can reach 2-4% [6, 7].
The Thalassemia International Federation (TIF) has recommended MCV as a crucial criterion for determining probable thalassemia carriers [15]. Carrier status was indicated by < 78 fL MCV and the presence of microcytic, hypochromic, and anisopoikilocytosis in peripheral blood smear [16]. This advice is supported by the Jordanian recommendations, which state that thalassemia should be further investigated in the general population if the MCV is < 80 fL [6]. Moreover, the association between MCV and thalassemia severity serves to support using MCV as an initial screening tool [8, 13]. Rund et al found a clear correlation between MCV and thalassemia variation severity, with β0 carriers exhibiting substantially lower MCV levels than β+ heterozygotes [17].
Sari et al and Rund et al found that recognizing the hematological diversity among β-thalassemia heterozygotes is important, depending on the type of mutation. Various MCV cutoffs may be more useful for particular demographics or geographical areas [3, 17]. For example, in South Asia, < 74 fL MCV is a good screening cutoff for women with prenatal anemia [18]. In Iran, < 27 pg MCH is more effective than in identifying β-thalassemia characteristics < 80 fL MCV [19].
Another hematological parameter that shows variation between different β-thalassemia phenotypes isboth Hb-A2 and Hb-F levels. Sari et al documented the differences in Hb-A2 and Hb-F levels between β0-thalassemia and β+-thalassemia. The Hb-A2 levels in patients with β0-thalassemia varied from 1.7% to 6.6% (median 5.1%). The low readings were compared to the cutoff point established by the screening recommendations for thalassemia, which were 2.9-6% (median 4.3%) for ββ+-thalassemia and 4-9% for thalassemia suggestive of β0 heterozygotes [3].
Furthermore, Zhang et al determined a new cutoff value (Hb-A2 ≥ 4.0%) for the diagnosis of β-thalassemia in Southwest China, which may be used to identify people more precisely [20].
Conclusion
Taken together, we found that Hb, MCV, MCH, and Hb-A2 can serve as effective screening markers to distinguish between β0-and β+-thalassemia variants in populations of carriers consisting of relatives of patients with thalassemia in Jordan. Additionally, DNA analysis is crucial to confirm our results and accurately identify whether the phenotype corresponds to β0- or β+-thalassemia, providing a more precise diagnosis and classification.
While these data support β-thalassemia phenotype classification and correlation, further research is needed to replicate these findings in a larger population. Specifically, studies should include more participants from Jordan, as well as other Mediterranean and Middle Eastern regions, to enhance the generalizability and applicability of the results.
Acknowledgments
We thank the members and management of the BioLab Medical Laboratory for their assistance and contributions to this work.
Financial Disclosure
This study funding was from the deanship of scientific research at Al-Balqa Applied University Salt, Jordan.
Conflict of Interest
The authors declare that they have no conflict of interest.
Informed Consent
Consent was obtained from all adult participants and legal guardians for participants under 18 years of age in this study.
Author Contributions
Ahmad Al Tibi and Amid Abdelnour collected blood samples and performed their analysis, followed by data collection and analysis. Diya Hasan, Ola Al-Sanabra, and Misaa Shawagfeh participated in study management and article writing. Ghiath Mansour, Moath Alqaraleh, and Tareq AlRamadneh performed statistical analysis. Mutaz Al-Khreisat helped in table’s preparation. George Burghel assisted with the article revision and data analysis.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
CBC: complete blood count; Hb: hemoglobin; IDA: iron deficiency anemia; MCH: mean corpuscular hemoglobin; MCV: mean corpuscular volume; PCR: polymerase chain reaction; TIF: Thalassemia International Federation
| References | ▴Top |
- Sadiq MF, Eigel A, Horst J. Spectrum of beta-thalassemia in Jordan: identification of two novel mutations. Am J Hematol. 2001;68(1):16-22.
doi pubmed - WHO, Regional desk review of haemoglobinopathies with an emphasis on thalassaemia and accessibility and availability of safe blood and blood products as per these patients’ requirement in South-East Asia Under Universal Health Coverage. 2021. [Online]. Available: http://apps.who.int/.
- Sari DP, Wahidiyat PA, Setianingsih I, Timan IS, Gatot D, Kekalih A. Hematological parameters in individuals with beta thalassemia trait in South Sumatra, Indonesia. Anemia. 2022;2022:3572986.
doi pubmed - Faraon R, Daraghmah M, Samarah F, Srour MA. Molecular characterization of beta-thalassemia intermedia in the West Bank, Palestine. BMC Hematol. 2019;19:4.
doi pubmed - Ebrahimi M, Mohammadi-asl J, Rahim F. Molecular spectrum and distribution of hemoglobinopathies in southwest of Iran: a seven-year retrospective study. J Hematopathol. 2020;13:97-103.
doi - Taani DN, Alabbadi DSDDQSDI, Nimri DO, Mahmoud DR, Hjazeen MR, Ishaq DA. Health & economic burden of b-thalassemia in Jordan for 2019. 2019; p. 34.
- Hasan D, Al Tibi A, Burghel G, Abdelnour A. Determining the current prevalence of beta-thalassemia variants in Jordan. Arch Med Sci. 2023;19(2):523-527.
doi pubmed - Shamoon RP, Al-Allawi NA, Cappellini MD, Di Pierro E, Brancaleoni V, Granata F. Molecular basis of beta-thalassemia intermedia in erbil province of Iraqi Kurdistan. Hemoglobin. 2015;39(3):178-183.
doi pubmed - Gharaibeh NS, Al-Sheyyab M, Batieha A. Detection of β-thalassemia carriers in Jordan. Ann Saudi Med. 1998;18(4):360-362.
doi - Alshorman A, Maghayreh M, Al Shorman A, Lafi M. The impact of mean corpuscular volume in identifying thalassemia trait through the premarital screening program in Northern Jordan. Jordan Med J. 2010;44(4):476-480.
- Basak AN. The molecular pathology of beta-thalassemia in Turkey: the Bogazici university experience. Hemoglobin. 2007;31(2):233-241.
doi pubmed - Asadov C, Abdulalimov E, Mammadova T, Gafarova S, Guliyeva Y, Aliyeva G. Genotype-phenotype correlations of beta-thalassemia mutations in an Azerbaijani population. Turk J Haematol. 2017;34(3):258-263.
doi pubmed - De Sanctis V, Kattamis C, Canatan D, Soliman AT, Elsedfy H, Karimi M, Daar S, et al. beta-Thalassemia distribution in the old world: an ancient disease seen from a historical standpoint. Mediterr J Hematol Infect Dis. 2017;9(1):e2017018.
doi pubmed - Mankhemthong K, Phusua A, Suanta S, Srisittipoj P, Charoenkwan P, Sanguansermsri T. Molecular characteristics of thalassemia and hemoglobin variants in prenatal diagnosis program in northern Thailand. Int J Hematol. 2019;110(4):474-481.
doi pubmed - Alassaf A, et al. 4th edition 2021 guidelines for the management of transfusion dependent thalassaemia (TDT). Thalass Int Fed. 2021; p. 1-351.
- Mehdi SR, Al Dahmash BA. A comparative study of hematological parameters of alpha and beta thalassemias in a high prevalence zone: Saudi Arabia. Indian J Hum Genet. 2011;17(3):207-211.
doi pubmed - Rund D, Filon D, Strauss N, Rachmilewitz EA, Oppenheim A. Mean corpuscular volume of heterozygotes for beta-thalassemia correlates with the severity of mutations. Blood. 1992;79(1):238-243.
pubmed - Baliyan M, Kumar M, Nangia A, Parakh N. Can RBC indices be used as screening test for beta-thalassemia in Indian antenatal women? J Obstet Gynaecol India. 2019;69(6):495-500.
doi pubmed - Karimi M, Rasekhi AR. Efficiency of premarital screening of beta-thalassemia trait using MCH rather than MCV in the population of Fars Province, Iran. Haematologia (Budap). 2002;32(2):129-133.
doi pubmed - Zhang J, He J, Mao X, Zeng X, Chen H, Su J, Zhu B. Haematological and electrophoretic characterisation of beta-thalassaemia in Yunnan province of Southwestern China. BMJ Open. 2017;7(1):e013367.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.