| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website https://jocmr.elmerjournals.com |
Review
Volume 18, Number 3, March 2026, pages 157-167

Advances in the Research of Hypoxia-Inducible Factor-1Alpha and Plasminogen Activator Inhibitor-1 in Vascular-Related Diseases
Xiao Xua, b, Xiao Hu Geb, c
aXinjiang Medical University Graduate School, Urumqi, Xinjiang Uygur Autonomous Region, Urumqi 831500, China
bDepartment of Vascular Surgery, People’s Hospital of Xinjiang Uygur Autonomous Region, Urumqi, Xinjiang Uygur Autonomous Region, Urumqi 831500, China
cCorresponding Author: Xiao Hu Ge, Department of Vascular Surgery, People’s Hospital of Xinjiang Uygur Autonomous Region, Urumqi, Xinjiang Uygur Autonomous Region, Urumqi 831500, China
Manuscript submitted July 21, 2025, accepted January 10, 2026, published online March 26, 2026
Short title: HIF-1α and PAI-1 in Vascular-Related Diseases
doi: https://doi.org/10.14740/jocmr6330
| Abstract | ▴Top |
Hypoxia-inducible factor-1α (HIF-1α) and plasminogen activator inhibitor-1 (PAI-1) play crucial roles in vascular homeostasis and pathological remodeling. Their intertwined regulatory network represents a focal point in vascular disease research. Under physiological conditions, HIF-1α and PAI-1 act coordinately to maintain vascular adaptability and repair capacity. However, under pathological conditions such as hypoxia or metabolic dysregulation, their aberrant activation constitutes a significant driver of vascular pathologies, including thrombosis, fibrosis, and atherosclerosis. Consequently, targeting this regulatory network may offer novel therapeutic approaches for vascular diseases. Cellular exposure to stressors such as hypoxia or inflammation induces the expression of key regulatory factors including HIF-1α and PAI-1. As the principal transcription factor mediating hypoxic responses, HIF-1α activates downstream target genes such as vascular endothelial growth factor (VEGF) and glucose transporter 1 (GLUT1), thereby regulating angiogenesis, metabolic reprogramming, and inflammatory responses. PAI-1, a serine protease inhibitor, contributes critically to thrombosis, fibrosis, and endothelial dysfunction through inhibition of the fibrinolytic system and modulation of extracellular matrix (ECM) degradation.
Keywords: HIF-1α; PAI-1; Vascular remodeling; Hypoxia signal; Fibrinolysis system; Targeted therapy
| Introduction | ▴Top |
Vascular diseases exhibit high global prevalence with continuously rising incidence, placing substantial economic burdens on societies and families. Consequently, a comprehensive investigation into the relationship between vascular diseases and hypoxia-inducible factor-1α (HIF-1α) and plasminogen activator inhibitor-1 (PAI-1) is crucial for advancing research in this field and establishing a theoretical foundation for understanding their pathogenesis.
The research landscape of the HIF-1α/PAI-1 axis is built upon pivotal historical discoveries. The field was inaugurated by the seminal identification of HIF-1 in the early 1990s, which established the molecular basis for oxygen sensing [1]. Subsequent research throughout the following decades elucidated HIF-1α’s central role in vascular biology, including angiogenesis and metabolic adaptation [2]. In parallel, the critical pathological function of PAI-1 as a key regulator of fibrinolysis and fibrosis in cardiovascular contexts was firmly established [3]. It is the more recent convergence of these two research trajectories that has spotlighted their intricate interplay within the hypoxic microenvironment as a major focus for understanding vascular disease mechanisms.
HIF-1α, a core transcriptional regulator in the cellular response to hypoxic microenvironments, exhibits biologically significant interactions with PAI-1 in the regulation of vascular homeostasis. In recent years, the molecular mechanisms underlying the interplay between HIF-1α and PAI-1, along with their synergistic roles in regulating the pathophysiology of various diseases, have become a major research focus. This article systematically reviews the structural and functional characteristics of the HIF-1α/PAI-1 axis and its molecular associations with vascular diseases, aiming to provide a solid theoretical basis for elucidating their pathological mechanisms.
Concept and function of HIF-1α
The molecular characteristics and functional mechanisms of HIF-1 were first elucidated by Semenza’s research team in 1992. HIF-1 functions as a heterodimer, comprising an oxygen-sensitive α subunit (HIF-1α, ∼120 kDa) and a constitutively expressed β subunit (HIF-1β, ∼91-94 kDa), with its biological activity primarily regulated by the α subunit [4]. Both subunits contain a conserved basic helix-loop-helix (bHLH) domain essential for DNA binding [5]. Research confirms that HIF-1α directly regulates a cluster of target genes—including erythropoietin (EPO), vascular endothelial growth factor (VEGF), and glucose transporter 1 (GLUT1)—all harboring conserved HIF-1α binding motifs in their regulatory regions. This observation establishes HIF-1α as a pivotal regulator within the hypoxia-adaptive transcriptional network. By orchestrating the reprogramming of multiple gene expression profiles, HIF-1α mediates critical physiological and pathological processes such as cellular metabolism, angiogenesis, and oxygen homeostasis, underscoring its central role in hypoxic signaling [6]. Furthermore, HIF-1α integrates oxygen-sensing pathways with epigenetic regulatory mechanisms to coordinate transcriptional reprogramming of glycolytic metabolism, angiogenesis, and erythropoiesis, thereby playing a core role in ischemic injury repair, tumor microenvironment adaptation, and oxygen homeostasis maintenance. Critically, the oxygen-dependent degradation domain (ODD) of HIF-1α and its interaction with prolyl hydroxylase (PHD) solidify its function as a molecular switch for dynamic oxygen signaling regulation.
Concept and function of PAI-1
PAI-1 is a serine protease inhibitor encoded by the SERPINE1 gene. It acts as the primary inhibitor of both urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA), thereby inhibiting fibrinolysis. PAI-1 is a single-chain glycoprotein with a molecular weight of approximately 45 kDa, composed of 379–381 amino acid residues. It contains nine α-helices and three β-sheet structures. The functional domains of PAI-1 include the reactive center loop (RCL), which mediates binding to plasminogen activators; a flexible hinge region involving helices D (hD), E (hE), and F (hF) that binds to vitronectin; and regions hD and hE, which also participate in the binding of low-density lipoprotein receptor-related protein (LRP) [7]. Depending on its source and characteristics, PAI can be classified into several types, including endothelial PAI (PAI-1), placental PAI (PAI-2), urinary PAI (PAI-3), and protease-nexin-1 (PAI-4), among which PAI-1 plays the most critical role in physiological processes [8]. PAI-1 is primarily secreted into the plasma in its active form, and its physiological functions are mainly manifested in the following aspects: by inhibiting the activities of uPA and tPA, it affects various cellular functions such as nerve growth, collagen activation, and tissue repair; during inflammatory responses, complement activation, and coagulation processes, PAI-1 exerts important inhibitory and degradative effects on relevant proteins; it protects cell membrane helices from degradation by plasma enzymes; it maintains cell-to-cell contact surfaces, thereby ensuring tissue integrity; and within the cell cycle, the intracellular accumulation of active PAI-1 and changes in its transcription are crucial for maintaining cell morphology, cell proliferation, cell adhesion to the matrix, signal transduction, and gene expression [9].
Relationship between HIF-1α and PAI-1
HIF-1α and PAI-1 engage in complex interregulatory mechanisms across various pathophysiological processes. Under hypoxic conditions, HIF-1α binds to the hypoxia response element (HRE) within the PAI-1 gene promoter, thereby activating PAI-1 transcription [10]. For instance, hypoxia stabilizes HIF-1α within the supernatant of alveolar macrophages and enhances its induction of PAI-1 [11]. Conversely, PAI-1 provides indirect feedback regulation of HIF-1α activity by modulating extracellular matrix (ECM) remodeling and cellular signaling pathways. Specifically, PAI-1 inhibits uPA and tPA, reducing plasmin generation. This, in turn, diminishes ECM degradation and promotes fibrin deposition, fostering a hypoxic microenvironment that activates HIF-1α [12]. The synergistic effects of HIF-1α and PAI-1 are particularly evident in pathological contexts. During tumor progression, hypoxia upregulates PAI-1 via HIF-1α, promoting tumor angiogenesis [13]. In thrombosis, HIF-1α-mediated hypoxic upregulation of PAI-1 may contribute to atherosclerotic plaque stability and increases the risk of thrombus formation [14]. Experimental studies demonstrate that hypoxia enhances HIF-1α stability and activity, significantly inducing PAI-1 expression in human hepatocellular carcinoma cells (HepG2 cells) and elucidating the mechanism underlying hypoxia-induced PAI-1 upregulation. Within this mechanism, HIF-1α and PAI-1 act synergistically: PAI-1 inhibits the fibrinolytic process, reducing fibrinolysis and promoting thrombus formation [15]. The synergistic regulatory interplay between HIF-1α and PAI-1 is summarized in Figure 1.
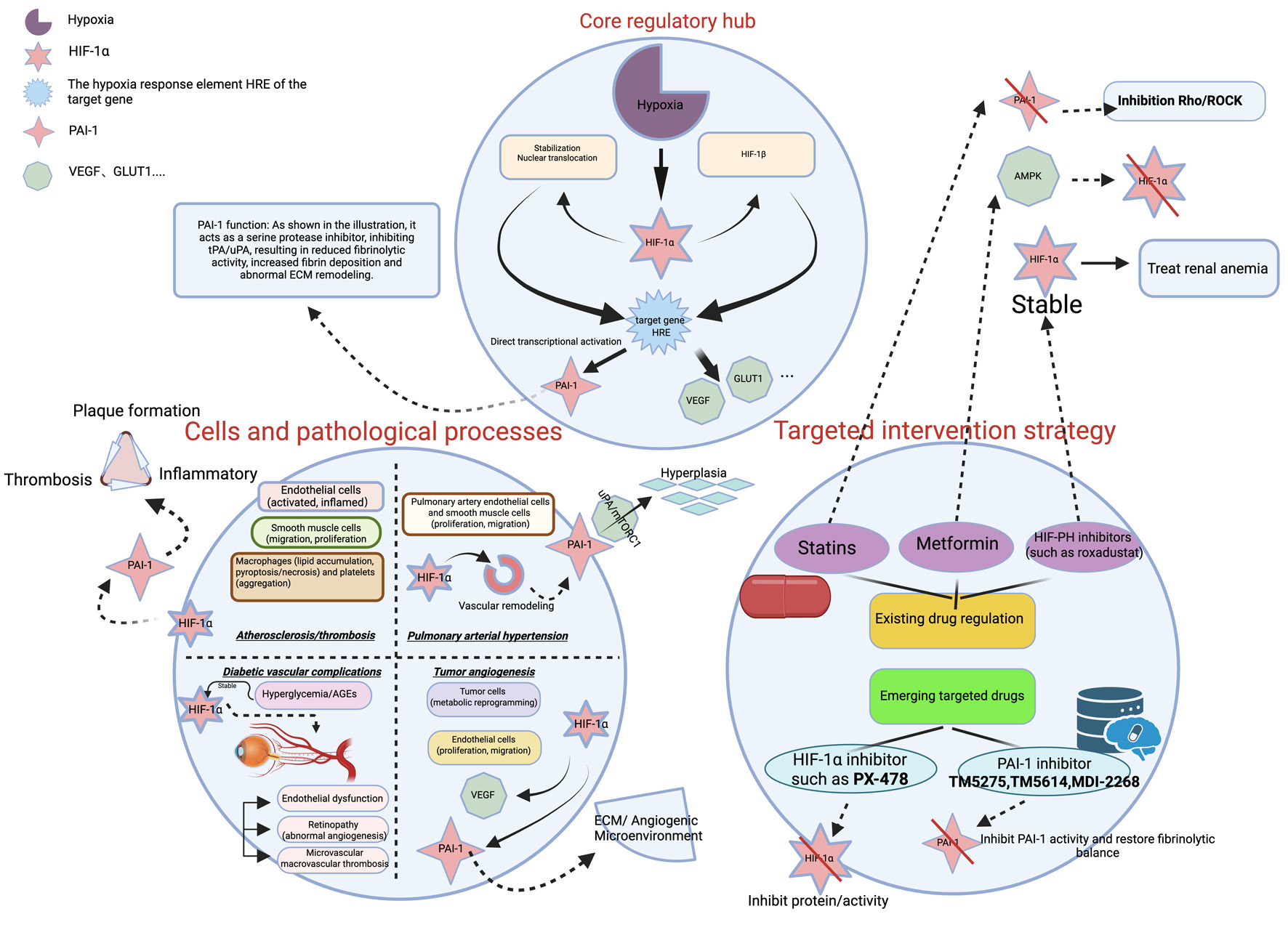 Click for large image | Figure 1. The HIF-1α/PAI-1 regulatory network in vascular diseases. Under hypoxic conditions, HIF-1α is stabilized and translocates to the nucleus, where it transcriptionally activates target genes including PAI-1, VEGF, and GLUT1 by binding to hypoxia response elements (HREs). Elevated PAI-1 contributes to the pathogenesis of various vascular disorders—such as atherosclerosis/thrombosis, pulmonary arterial hypertension (PAH), diabetic vascular complications, and tumor angiogenesis—through mechanisms involving fibrinolysis inhibition, inflammatory modulation, extracellular matrix (ECM) remodeling, and vascular remodeling. The diagram also summarizes current targeted intervention strategies, including existing drugs (e.g., statins) and emerging inhibitors targeting HIF-1α (e.g., PX-478) or PAI-1 (e.g., TM5275, TM5614, MDI-2268). HIF-1α: hypoxia-inducible factor-1α; PAI-1: plasminogen activator inhibitor-1. VEGF: vascular endothelial growth factor; GLUT1: glucose transporter 1. |
| The Role of HIF-1α and PAI-1 in Vascular Diseases | ▴Top |
The multifaceted roles of HIF-1α and PAI-1 in atherosclerosis
The pathogenesis of atherosclerosis involves a complex interplay of molecular signals, with HIF-1α and PAI-1 serving as pivotal, interconnected regulators. The process is initiated at sites predisposed to plaque formation, such as arterial branches characterized by low shear stress. Under these conditions, endothelial HIF-1α expression is upregulated via transcriptional activation by nuclear factor-κB (NF-κB) and protein stabilization mediated by the deubiquitinase Cezanne (also known as OTUD7B). This early HIF-1α activation drives endothelial metabolic reprogramming and inflammatory responses, establishing a pro-atherogenic milieu [16].
Subsequent hypoxia within the evolving plaque further amplifies HIF-1α signaling, which orchestrates key cellular events. In vascular smooth muscle cells (VSMCs), HIF-1α directly binds to the promoter of the Kruppel-like factor 4 (KLF4) gene, inducing its expression. This HIF-1α-mediated KLF4 upregulation activates pro-migratory signaling pathways, enhancing VSMC migration—a critical step in vascular remodeling and neointima formation [17]. Concurrently, within macrophages, HIF-1α activation shifts cellular metabolism by reducing oxidative phosphorylation and ATP levels while elevating reactive oxygen species (ROS), thereby promoting necroptosis. This process is fine-tuned by HIF-1α through the upregulation of miR-210 and downregulation of miR-383, with the latter targeting poly(ADP-ribose) glycohydrolase (PARG) to influence DNA damage repair pathways, collectively affecting plaque cellularity and stability [18]. The therapeutic relevance of targeting macrophage HIF-1α is underscored by the action of salidroside (a bioactive compound derived from Rhodiola rosea), a natural compound that ameliorates macrophage lipid accumulation and reduces atherosclerotic plaques by inhibiting HIF-1α-induced pyroptosis [19].
In parallel, PAI-1 expression is markedly elevated in atherosclerotic lesions and is functionally linked to inflammatory pathways (e.g., NF-κB signaling) and oxidative stress [20]. PAI-1 contributes to disease progression through multiple mechanisms: it promotes ox-LDL-induced senescence in aortic endothelial cells and macrophages via p16/p21 and p53-mediated cell cycle arrest [21]; its genetic deficiency or pharmacological inhibition in animal models significantly reduces lesion size by dampening macrophage infiltration, inhibiting VSMC proliferation/migration, and mitigating oxidative stress [22]. The anti-atherosclerotic effects of agents like baicalein are partly attributable to the inhibition of PAI-1 activity, leading to attenuated endothelial damage and suppressed thrombosis [20].
A critical mechanistic link in atherosclerosis is the HIF-1α-mediated upregulation of PAI-1. Within the hypoxic plaque microenvironment, HIF-1α activation directly increases the expression of pro-thrombotic factors, including PAI-1 [14]. This regulatory relationship, modulated by hemodynamic forces, results in suppressed fibrinolysis and accelerated thrombotic complications [23]. The clinical correlation of this pathway is evidenced by the increased presence of HIF-1α-immunopositive cells within coronary artery plaques, particularly in areas of thrombosis [14]. The interconnected nature of the HIF-1α and PAI-1 pathways is further highlighted by the action of silymarin, which attenuates atherogenesis by inhibiting HIF-1α-mediated pyroptosis [19]. In summary, HIF-1α and PAI-1 form a synergistic regulatory network that coordinates endothelial dysfunction, smooth muscle cell migration, macrophage inflammation and death, and ultimately thrombosis, driving the progression of atherosclerosis.
Pulmonary arterial hypertension (PAH)
HIF-1α drives pulmonary vascular remodeling and hypoxic adaptation
Research has established that calpain-1 exacerbates pulmonary vascular remodeling and fibrosis, thereby promoting the development of PAH. This occurs through NF-κB (p65)-mediated upregulation of HIF-1α expression under hypoxic conditions [24]. Hypoxia induces increased expression of connexin 43 (Cx43) and enhanced phosphorylation at its Ser368 site in both rat pulmonary arteries and pulmonary arterial smooth muscle cells (PASMCs). These changes promote PASMC proliferation and migration. The HIF-1α inhibitor actinomycin D attenuates this proliferative/migratory effect. Furthermore, chromatin immunoprecipitation assays confirmed direct binding of HIF-1α to the Cx43 promoter, demonstrating that HIF-1α acts as an upstream regulator enhancing Cx43 expression. Collectively, these findings indicate that the HIF-1α/Cx43 axis plays a critical role in hypoxia-induced PAH pathogenesis. Modulation of this axis may therefore represent a therapeutic strategy for controlling pulmonary vascular remodeling [25].
The unique role of PAI-1 deficiency in PAH pathogenesis
PAH patients exhibit deficient PAI-1 expression in the smooth muscle compartment of remodeled small pulmonary arteries (PAs) and in early-passage PASMCs compared to non-PAH controls. Both male and female PAI-1 knockout (PAI-1−/−) mice develop spontaneous pulmonary vascular remodeling and PAH, characterized by increased pulmonary arterial medial thickness, elevated systolic right ventricular pressure, and right ventricular hypertrophy [26]. Mechanistically, PAI-1 deficiency in PAH subjects potentiates excessive PASMC proliferation and vascular remodeling through unopposed uPA activity, driving mTORC1 and TGF-β signaling upregulation [27]. This deficiency is clinically relevant: PAH patients demonstrate significantly reduced serum and lung tissue PAI-1 levels alongside elevated uPA, correlating with disease progression [28].
Therapeutic outlook: targeting the hypoxia-fibrinolysis crosstalk
The contrasting roles of HIF-1α (promoter) and PAI-1 (potential suppressor) in PAH highlight the complexity of vascular remodeling. Future therapeutic strategies may need to consider the balance between hypoxic signaling and fibrinolytic activity. Simultaneous modulation of the HIF-1α pathway and restoration of local PAI-1 activity or its downstream effects could represent a novel approach to stabilizing the pulmonary vasculature.
Thrombosis
PAI-1: the core regulator of fibrinolytic balance and thrombotic risk
In acute cerebral infarction, serum anti-PAI-1 antibody levels are significantly elevated and independently associated with established vascular risk factors (age, sex, hypertension, diabetes, cardiovascular disease) and carotid intima-media thickness. Multivariate analysis confirms anti-PAI-1 antibodies as independent predictors of acute ischemic stroke [29]. Complementing this, a population-based nested case-control study established a dose-response relationship between plasma PAI-1 levels and future venous thromboembolism (VTE) risk. Notably, PAI-1 mediates approximately 15% of VTE risk in obese individuals, representing the first epidemiological evidence linking elevated PAI-1 to incident VTE [30]. Therapeutically, Danshen-Honghua injection ameliorates thrombotic risk by reducing PAI-1 levels and restoring fibrinolytic homeostasis, demonstrating both PAI-1’s mechanistic role in thrombosis and its potential as a therapeutic target for anticoagulation enhancement [31].
HIF-1α: shaping the pro-thrombotic pathological microenvironment
This regulatory axis extends to dermal thrombosis, where mechanical compression induces hypoxia-mediated HIF-1α activation and PAI-1 induction, potentiating local fibrin deposition. HIF-1 inhibitors abolish this prothrombotic phenotype while reducing PAI-1 transcription [32].
The hypoxia-HIF-1α-PAI-1 pathway: a common axis in thrombosis
A critical mechanistic link in atherothrombosis is the HIF-1α-mediated upregulation of PAI-1. Within hypoxic environments, such as an evolving atherosclerotic plaque or compressed tissue, HIF-1α activation directly increases the expression of pro-thrombotic factors, including PAI-1 [14, 23,]. This regulatory relationship results in suppressed fibrinolysis and accelerated thrombotic complications. In diabetic retinopathy, retinal hypoxia concurrently activates HIF-1α and PAI-1, which collectively promote thrombosis, pathological angiogenesis, and inflammation to drive disease progression. Thus, the hypoxia-HIF-1α-PAI-1 pathway serves as a fundamental mechanism connecting local tissue ischemia to systemic thrombotic risk across various vascular pathologies.
Diabetic vascular complications
PAI-1 mediates microvascular and macrovascular complications
Clinical investigations demonstrate elevated PAI-1 levels in both serum and tear fluid of diabetic retinopathy (DR) patients compared to diabetic controls without DR and healthy subjects. Notably, proliferative DR (PDR) patients exhibit significantly higher serum PAI-1 concentrations than non-proliferative DR (NPDR) cases, establishing a strong positive correlation between PAI-1 elevation and DR severity [33]. Complementary research implicates PAI-1 in the pathogenesis of cardiovascular complications in type 2 diabetes mellitus (T2DM). Insulin resistance stimulates PAI-1 overproduction, creating a prothrombotic state that accelerates vascular disease onset and exacerbates clinical severity [34].
HIF-1α in diabetic chronic hypoxia and inflammation
In the diabetic state, factors like advanced glycation end products (AGEs) and oxidative stress can stabilize HIF-1α even under normoxic conditions. This contributes to chronic low-grade inflammation and aberrant angiogenesis in tissues like the retina [33].
Synergistic aggravation of vascular injury: a vicious cycle
Hyperglycemia and hypoxia synergistically upregulate both HIF-1α and PAI-1. This creates a vicious cycle: PAI-1 promotes thrombosis and ischemia, which in turn enhances HIF-1α stability; activated HIF-1α further transactivates PAI-1 and other inflammatory mediators. This interconnected network accelerates endothelial dysfunction, vascular occlusion, and end-organ damage in diabetes.
Tumor angiogenesis
HIF-1α: the master transcriptional driver of tumor angiogenesis
In colorectal cancer, HIF-1α drives tumor angiogenesis by activating VEGF and other pro-angiogenic factors while promoting endothelial cell proliferation. Under hypoxia, HIF-1α further induces lactate uptake and stimulates angiogenesis-associated signaling pathways [35]. The ROS-ATM-CHK2 axis in the hypoxic tumor microenvironment enhances angiogenesis through HIF-1α stabilization. Specifically, CHK2 interacts with HIF-1α to inhibit its ubiquitination, thereby increasing HIF-1α stability and upregulating VEGF expression [36]. Conversely, β-hydroxybutyrate suppresses HIF-1α activity in colitis-associated tumor models, reducing angiogenic factor expression and inhibiting tumor-associated angiogenesis [37]. In TP53-mutated head and neck squamous cell carcinoma (HNSCC), HIF-1α activates the STAT3 pathway to induce VEGF expression, promoting tumor angiogenesis and lymphangiogenesis. Downregulation of HIF-1α and VEGF in this context inhibits these processes, reducing tumor recurrence and metastasis [38].
The complex role of PAI-1 in tumor angiogenesis and microenvironment remodeling
In cutaneous malignancies, elevated PAI-1 expression potentiates tumor angiogenesis through multifaceted mechanisms. These include: 1) inhibition of fibrinolytic activity generating pro-angiogenic fibrin matrices, 2) dysregulation of ECM remodeling, and 3) ligand-receptor interactions (e.g., with integrins) that collectively promote endothelial cell migration and neovascularization [39]. Contrastingly, in malignant pleural mesothelioma, PAI-1 exerts tumor-suppressive effects by restricting ECM remodeling and cellular migration, thereby inhibiting tumor progression independent of angiogenic stimuli [40]. Glioblastoma studies further reveal that cancer stemness marker ALDH1A3 drives angiogenesis via paracrine secretion of PAI-1 and IL-8 [41]. This evidence establishes PAI-1 as a context-dependent regulator of tumor vascularization.
Transcriptional activation of PAI-1 by HIF-1α: a key step in cooperative tumor vascularization
A critical functional interaction in tumors is the direct transactivation of PAI-1 by HIF-1α. Under hypoxic conditions, activated HIF-1α translocates to the nucleus and dimerizes with HIF-1β. This heterodimeric complex binds hypoxia response elements (HREs) in the PAI-1 promoter, directly transactivating PAI-1 transcription. Elevated PAI-1 suppresses proteolytic fibrinolytic activity, which consequently potentiates endothelial cell proliferation and migration, thereby facilitating tumor neovascularization [35]. This mechanism exemplifies a direct molecular synergy where HIF-1α amplifies the pro-angiogenic tumor microenvironment through PAI-1.
Dysregulation of HIF-1α and PAI-1 in infectious diseases
Systemic dysregulation and immunothrombosis in sepsis
Sepsis induces profound systemic hypoxia and a cytokine storm, leading to concurrent overexpression of PAI-1 in endothelial and immune cells. This synergistic upregulation promotes the formation of “immunothrombi,” contributing to disseminated intravascular coagulation (DIC) and multiple organ failure [42].
Endothelialitis and thrombotic complications in COVID-19
SARS-CoV-2 infection stabilizes hypoxia-inducible factor HIF-1α, which directly upregulates the expression of the viral receptor ACE2 in host cells, thereby enhancing viral entry and replication. Concurrently, activated HIF-1α drives a pro-inflammatory cytokine storm, exacerbating pulmonary tissue damage. The study demonstrates that targeting HIF-1α reduces viral load and inflammatory responses, highlighting its potential as a therapeutic strategy for severe COVID-19 [43].
| Clinical Relevance of HIF-1α and PAI-1: Impact of Age, Sex, and Comorbidities | ▴Top |
Impact of age
Aging represents a significant independent risk factor for vascular diseases. This process is characterized by a decline in HIF-1α signaling responsiveness and a systemic downregulation of genes associated with the mobilization and function of endothelial progenitor cells (EPCs). In a murine model of acute hindlimb ischemia, aged mice exhibited a delayed and significantly attenuated peak in both HIF-1α mRNA and protein expression within ischemic muscle tissue compared to young mice. This was accompanied by a corresponding reduction in the expression of its key downstream pro-angiogenic factor, VEGF. These findings suggest a blunted response of EPCs to hypoxia-induced HIF-1α activation in aged individuals, leading to diminished neovascularization capacity in ischemic tissues [44]. Concurrently, aging-associated chronic low-grade inflammation and vascular stress drive a sustained increase in PAI-1 expression. Experimental studies indicate that pathological stress, such as hypertension, can directly induce excessive vascular PAI-1 expression. PAI-1 itself acts as a key mediator driving vascular aging and inflammation. In an L-NAME-induced hypertensive mouse model, the PAI-1 antagonist TM5441 significantly attenuated blood pressure elevation, improved vasodilation function, and effectively inhibited the expression of vascular senescence markers (e.g., SA-β-gal activity, p53, and p21 proteins). This study provides direct evidence that PAI-1 is not merely a biomarker of aging and inflammation but a critical effector promoting the senescent phenotype of endothelial cells and vascular inflammation [45]. Elevated PAI-1 levels are closely associated with an increased risk of atherothrombotic events prevalent in the elderly population [46].
Impact of sex
Cardiovascular disease risk exhibits significant sexual dimorphism. The effects of estrogen display considerable complexity, depending on receptor subtypes and cellular context. Research has found that estrogen receptor β can significantly inhibit the transcriptional activity of HIF-1 and the expression of its downstream gene VEGF by downregulating ARNT, an essential cofactor for HIF-1 [47]. This suggests that, within specific pathways, estrogen signaling may suppress rather than promote hypoxic adaptive responses, revealing a more nuanced and bidirectional regulatory network in processes such as angiogenesis. Conversely, androgens may promote PAI-1 expression. Studies have shown that in prostate cancer tissues, the expression level of the androgen receptor (AR) is significantly positively correlated with the expression of HIF-1α, HIF-2α, and their key downstream effector, VEGF [48]. This indicates that AR signaling may enhance or synergize with HIF-α activity to promote tumor angiogenesis and adaptation to the hypoxic microenvironment.
Impact of common comorbidities
Metabolic diseases
Obesity is a potent stimulus for the overproduction of PAI-1. Research has found that in obese individuals, the mRNA expression level of PAI-1 in abdominal subcutaneous adipose tissue (but not in femoral fat) is significantly positively correlated with both plasma PAI-1 concentration and the degree of systemic insulin resistance. Importantly, following dietary intervention leading to an average weight loss of 9%, PAI-1 gene expression in abdominal subcutaneous fat significantly decreased, accompanied by improvements in metabolic parameters [49].
Hypertension
Persistent hypertension potently upregulates PAI-1 expression via the renin-angiotensin system, with angiotensin II serving as a key inducer in this process. Notably, individual genetic differences influence the PAI-1 responsiveness to angiotensin II. Studies indicate that hypertensive patients carrying the PAI-1 4G/4G genotype exhibit a more pronounced increase in PAI-1 levels upon angiotensin II stimulation [50]. This suggests that genetic predisposition and systemic factors jointly determine the elevated PAI-1 state and potential thrombotic risk in hypertensive patients.
| Clinical Applications and Translational Research | ▴Top |
Clinical applications and translational research of HIF-1α
HIF-1α represents a pivotal therapeutic target across multiple disease states. In oncology, HIF-1α overexpression correlates with solid tumor invasion, metastasis, chemo-radioresistance, and poor prognosis [51]. Pharmacological inhibitors or genetic silencing of HIF-1α suppress tumor angiogenesis and glycolytic metabolism [52]. Furthermore, HIF-1α mediates immune evasion through PD-L1 upregulation, suggesting potential synergy with immune checkpoint inhibitors [53]. For ischemic conditions, HIF-1α activators (e.g., roxadustat) ameliorate perfusion deficits in myocardial infarction and limb ischemia models by inducing VEGF and EPO expression [54]. Hypoxic preconditioning of mesenchymal stem cells enhances their paracrine-mediated angiogenic potential in ischemic tissues [55]. In chronic kidney disease, pharmacological HIF-1α stabilization improves renal anemia by stimulating erythropoiesis [56]. Additionally, HIF-1α modulates inflammatory pathogenesis through regulation of macrophage polarization states [57].
Clinical applications and translational research of PAI-1
PAI-1 represents a critical therapeutic target across thrombotic, fibrotic, oncologic, and metabolic pathologies. Elevated PAI-1 levels establish a hypercoagulable state, correlating with VTE, atherogenesis, and metabolic syndrome while serving as a prognostic biomarker for adverse outcomes [58]. In fibrotic disorders, PAI-1 drives pulmonary, hepatic, and renal fibrogenesis through matrix metalloproteinase (MMP) inhibition [59], with inhibitors like TM5275 demonstrating significant antifibrotic efficacy in preclinical models [60]. Within tumor microenvironments, PAI-1 facilitates obesity-associated oncogenesis by enhancing integrin-mediated collagen I (COL1) engagement and pathological ECM remodeling [61]. In metabolic diseases, pathologically elevated PAI-1 mediates adipose tissue fibrosis and chronic inflammation in obesity and type 2 diabetes mellitus, positioning it as a promising intervention target for metabolic syndrome [62]. Collectively, these findings establish PAI-1 as a multifunctional mediator in thrombosis, fibrosis, cancer progression, and metabolic dysregulation [63].
Regulatory strategies based on existing clinical drugs
Drugs targeting metabolic diseases
Metformin, a first-line medication for type 2 diabetes, exerts pleiotropic protective effects partly through modulating the HIF-1α signaling pathway. Studies have shown that in models of sepsis-associated lung injury, metformin effectively inhibits the accumulation of HIF-1α protein and its mediated aerobic glycolysis by activating the AMP-activated protein kinase (AMPK) pathway [64]. This mechanism suggests the potential of metformin, via AMPK regulation of the HIF-1α axis, to exert protective effects in hypoxic or inflammatory conditions.
Lipid-lowering and cardioprotective drugs
Beyond cholesterol reduction, statins exhibit pleiotropic properties. Clinical meta-analyses have confirmed that statin treatment significantly reduces plasma levels of PAI-1 [65], a mechanism potentially linked to the inhibition of Rho protein family signaling [66]. Furthermore, statins improve vascular function by suppressing the RhoA/ROCK pathway [67]. Theoretically, this pathway is also a key regulator of HIF-1α stability under hypoxic conditions, hinting at a potential additional mechanism underlying statin-induced plaque stabilization and improved prognosis.
Drugs targeting renal anemia
HIF prolyl hydroxylase inhibitors (e.g., roxadustat, vadadustat) stabilize HIF-α, mimicking the hypoxic response, and effectively stimulate endogenous EPO production. High-level evidence-based medical studies demonstrate that roxadustat shows significant efficacy in correcting hemoglobin levels in patients with chronic kidney disease-associated anemia, even surpassing traditional erythropoiesis-stimulating agents [68]. However, a critical question requiring long-term clinical monitoring is whether the systemic elevation of HIF-α levels by these drugs may potentially influence PAI-1 expression and thrombotic risk.
Emerging therapeutic agents targeting HIF-1α and PAI-1
Agents targeting HIF-1α
Direct pharmacological inhibition of HIF-1α has been pursued as a strategy to disrupt the adaptive responses of tumors and other diseases to hypoxia.
PX-478: A potent and selective small-molecule inhibitor of HIF-1α [69]. It functions by reducing HIF-1α protein levels and inhibiting its transcriptional activity, independent of tumor suppressor genes like VHL and p53 [70]. Preclinical studies have demonstrated its efficacy in sensitizing various solid tumors to other therapies. Notably, recent research highlights that PX-478 can overcome HIF-1α-driven resistance to a novel form of cell death (cuproptosis) in solid tumors [71]. Beyond oncology, PX-478 has shown promise in preserving pancreatic β-cell function in mouse models of diabetes, suggesting potential therapeutic repurposing [72]. While it has entered phase I clinical trials for advanced cancers, further development has faced challenges.
Agents targeting PAI-1
Inhibiting PAI-1 activity aims to restore fibrinolytic balance and counteract its pathological roles in fibrosis and tumor progression.
TM5275: An orally bioavailable small-molecule PAI-1 inhibitor [73]. It has been shown to enhance fibrinolysis on vascular endothelial cells by preventing PAI-1 from complexing with tPA [74].
TM5614: Another oral PAI-1 inhibitor under investigation. Preclinical evidence suggests that it can reverse resistance to PD-1 immunotherapy in non-small cell lung cancer models, and it is currently being evaluated in a phase II clinical trial (NCT05015960) [75].
MDI-2268: A potent small-molecule PAI-1 inhibitor effective both in vitro and in vivo [76]. Studies demonstrate that in disease models, MDI-2268, as a PAI-1 inhibitor, can promote a shift in the immune microenvironment towards a pro-inflammatory state, thereby proving its bioactivity and therapeutic potential in modulating inflammatory responses [77].
Challenges and directions in translational research
HIF-1α demonstrates context-dependent duality: in ischemic pathologies, its activation promotes angiogenesis and cytoprotection, whereas in tumor microenvironments it facilitates oncogenic progression. This necessitates development of tissue-targeted delivery systems to spatially regulate its beneficial versus detrimental effects. Similarly, PAI-1 exhibits functional dichotomy, promoting hypercoagulable states in thrombosis (VTE, atherosclerosis, metabolic syndrome) while potentially conferring hemostatic protection in bleeding disorders. Such dualistic pathophysiology mandates precise patient stratification for therapeutic interventions. Current translational research focuses on integrating cutting-edge multi-omics approaches, including single-cell sequencing, spatial transcriptomics, and AI-driven predictive modeling, to deconvolute the HIF-1α/PAI-1 regulatory networks and optimize personalized therapeutic strategies. However, clinical translation of HIF-1α/PAI-1-targeted agents faces significant challenges: limited bioavailability, off-target effects, and unresolved long-term safety concerns remain critical barriers [78]. Systematic resolution of these limitations is essential for clinical implementation of effective targeted therapies.
| Challenges and Future Directions | ▴Top |
Current mechanistic research on HIF-1α and PAI-1 faces notable constraints. Tissue-specific heterogeneity and dynamic changes across disease stages limit the generalizability of findings, as both factors exhibit context-dependent functional variation. Moreover, interspecies discrepancies between animal models and human pathophysiology restrict translational relevance, compounding research complexity. Regarding therapeutic translation, HIF-1α/PAI-1-targeted agents encounter significant barriers including off-target effects and safety concerns, necessitating further pharmacological optimization to mitigate adverse events. Concurrently, biomarker validation requires multicenter cohort studies to establish robust clinical applicability. Emerging technologies present transformative opportunities in this field. Single-cell resolution analyses can delineate cell subtype-specific regulatory networks, elucidating spatiotemporal mechanisms of HIF-1α and PAI-1. Artificial intelligence-assisted drug design coupled with computational patient stratification enhances therapeutic precision and development efficiency. Strategic integration of these approaches holds promise to: 1) propel fundamental understanding of HIF-1α/PAI-1 biology, 2) address existing translational challenges, and 3) expedite clinical implementation of targeted interventions.
Acknowledgments
Technical support provided by the Laboratory of Xinjiang Medical University graduate school is gratefully acknowledged. The authors also appreciate the insightful discussions with their colleagues.
Financial Disclosure
This research was supported by the following two projects: Department of Science and Technology of Xinjiang Uygur Autonomous Region, Grant No. 2022D01C633.
Conflict of Interest
The authors have no conflict of interest to declare.
Author Contributions
Xiao Xu: conceptualization, methodology, investigation, data curation, and writing – original draft. Xiao Hu Ge: conceptualization, methodology, supervision, writing – review & editing.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Semenza GL. Surviving ischemia: adaptive responses mediated by hypoxia-inducible factor 1. J Clin Invest. 2000;106(7):809-812.
doi pubmed - Wu D, Huang RT, Hamanaka RB, Krause M, Oh MJ, Kuo CH, Nigdelioglu R, et al. HIF-1alpha is required for disturbed flow-induced metabolic reprogramming in human and porcine vascular endothelium. Elife. 2017;6.
doi pubmed - Fay WP. Plasminogen activator inhibitor 1, fibrin, and the vascular response to injury. Trends Cardiovasc Med. 2004;14(5):196-202.
doi pubmed - Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12(12):5447-5454.
doi pubmed - Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510-5514.
doi pubmed - Zhu P, Tang M, Yan D. Research progress of HIF-1α and its related signal tranuction pathways in diseases. Chinese Journal of Immunology. 2020;36(13):1650-1657.
- McMahon B, Kwaan HC. The plasminogen activator system and cancer. Pathophysiol Haemost Thromb. 2008;36(3-4):184-194.
doi pubmed - Morrow GB, Mutch NJ. Past, present, and future perspectives of plasminogen activator inhibitor 1 (PAI-1). Semin Thromb Hemost. 2023;49(3):305-313.
doi pubmed - Sillen M, Declerck PJ. A narrative review on plasminogen activator inhibitor-1 and its (Patho)physiological role: to target or not to target? Int J Mol Sci. 2021;22(5):2721.
doi pubmed - Loboda A, Jozkowicz A, Dulak J. HIF-1 and HIF-2 transcription factors—similar but not identical. Mol Cells. 2010;29(5):435-442.
doi pubmed - Jeny F, Bernaudin JF, Valeyre D, Kambouchner M, Pretolani M, Nunes H, Planes C, et al. Hypoxia promotes a mixed inflammatory-fibrotic macrophages phenotype in active sarcoidosis. Front Immunol. 2021;12:719009.
doi pubmed - Gawronska-Kozak B, Machcinska-Zielinska S, Walendzik K, Kopcewicz M, Paakkonen M, Wisniewska J. Hypoxia and Foxn1 alter the proteomic signature of dermal fibroblasts to redirect scarless wound healing to scar-forming skin wound healing in Foxn1(-/-) mice. BMC Biol. 2024;22(1):193.
doi pubmed - Jung SY, Song HS, Park SY, Chung SH, Kim YJ. Pyruvate promotes tumor angiogenesis through HIF-1-dependent PAI-1 expression. Int J Oncol. 2011;38(2):571-576.
doi pubmed - Matsuura Y, Yamashita A, Iwakiri T, Sugita C, Okuyama N, Kitamura K, Asada Y. Vascular wall hypoxia promotes arterial thrombus formation via augmentation of vascular thrombogenicity. Thromb Haemost. 2015;114(1):158-172.
doi pubmed - Sanagawa A, Iwaki S, Asai M, Sakakibara D, Norimoto H, Sobel BE, Fujii S. Sphingosine 1-phosphate induced by hypoxia increases the expression of PAI-1 in HepG2 cells via HIF-1alpha. Mol Med Rep. 2016;14(2):1841-1848.
doi pubmed - Feng S, Bowden N, Fragiadaki M, Souilhol C, Hsiao S, Mahmoud M, Allen S, et al. Mechanical activation of hypoxia-inducible factor 1alpha drives endothelial dysfunction at atheroprone sites. Arterioscler Thromb Vasc Biol. 2017;37(11):2087-2101.
doi pubmed - Shan F, Huang Z, Xiong R, Huang QY, Li J. HIF1alpha-induced upregulation of KLF4 promotes migration of human vascular smooth muscle cells under hypoxia. J Cell Physiol. 2020;235(1):141-150.
doi pubmed - Karshovska E, Wei Y, Subramanian P, Mohibullah R, Geissler C, Baatsch I, Popal A, et al. HIF-1alpha (Hypoxia-Inducible Factor-1alpha) promotes macrophage necroptosis by regulating miR-210 and miR-383. Arterioscler Thromb Vasc Biol. 2020;40(3):583-596.
doi pubmed - Guo W, Huang R, Bian J, Liao Q, You J, Yong X, Wang Y, et al. Salidroside ameliorates macrophages lipid accumulation and atherosclerotic plaque by inhibiting Hif-1alpha-induced pyroptosis. Biochem Biophys Res Commun. 2025;742:151104.
doi pubmed - Zhang X, Qin Y, Ruan W, Wan X, Lv C, He L, Lu L, et al. Targeting inflammation-associated AMPK//Mfn-2/MAPKs signaling pathways by baicalein exerts anti-atherosclerotic action. Phytother Res. 2021;35(8):4442-4455.
doi pubmed - Liang X, Zhang J, Yu J, Zhao J, Yang S. Quercetin ameliorates ox-LDL-induced cellular senescence of aortic endothelial cells and macrophages by p16/p21, p53/SERPINE1, and AMPK/mTOR pathways. Eur J Med Res. 2025;30(1):359.
doi pubmed - Khoukaz HB, Ji Y, Braet DJ, Vadali M, Abdelhamid AA, Emal CD, Lawrence DA, et al. Drug targeting of plasminogen activator inhibitor-1 inhibits metabolic dysfunction and atherosclerosis in a murine model of metabolic syndrome. Arterioscler Thromb Vasc Biol. 2020;40(6):1479-1490.
doi pubmed - Li S, Xu Z, Wang Y, Chen L, Wang X, Zhou Y, Lei D, et al. Recent advances of mechanosensitive genes in vascular endothelial cells for the formation and treatment of atherosclerosis. Genes Dis. 2024;11(3):101046.
doi pubmed - Deng H, Tian X, Sun H, Liu H, Lu M, Wang H. Calpain-1 mediates vascular remodelling and fibrosis via HIF-1alpha in hypoxia-induced pulmonary hypertension. J Cell Mol Med. 2022;26(10):2819-2830.
doi pubmed - Han XJ, Zhang WF, Wang Q, Li M, Zhang CB, Yang ZJ, Tan RJ, et al. HIF-1alpha promotes the proliferation and migration of pulmonary arterial smooth muscle cells via activation of Cx43. J Cell Mol Med. 2021;25(22):10663-10673.
doi pubmed - Kudryashova TV, Zaitsev SV, Jiang L, Buckley BJ, McGuckin JP, Goncharov D, Zhyvylo I, et al. PAI-1 deficiency drives pulmonary vascular smooth muscle remodeling and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2024;327(3):L319-L326.
doi pubmed - Kudryashova TV, Zaitsev S, Jiang L, Buckley BJ, McGuckin JP, Goncharov D, Zhyvylo I, et al. PAI-1 deficiency drives pulmonary vascular smooth muscle remodeling and pulmonary hypertension. bioRxiv. 2023.
doi pubmed - Shoji M, Matsui T, Tanaka H, Nomura K, Tsujita H, Kodama Y, Koba S, et al. Fibrinolytic markers could be useful predictors of severity in patients with pulmonary arterial hypertension: a retrospective study. Thromb J. 2021;19(1):78.
doi pubmed - Kubota M, Yoshida Y, Kobayashi E, Matsutani T, Li SY, Zhang BS, Mine S, et al. Serum anti-SERPINE1 antibody as a potential biomarker of acute cerebral infarction. Sci Rep. 2021;11(1):21772.
doi pubmed - Greenberg IM. Intraoperative ultrasonography with a cystoscope for the biopsy of a deep-seated brain lesion: case study. Neurosurgery. 1986;19(1):49-58.
doi pubmed - Yin W, Li J, Han Z, Wang S, Wu F, Yu C, Yan X, et al. Effect of Danhong injection on pharmacokinetics and pharmacodynamics of rivaroxaban in rats. Naunyn Schmiedebergs Arch Pharmacol. 2025;398(4):3617-3629.
doi pubmed - Kaneko M, Minematsu T, Yoshida M, Nishijima Y, Noguchi H, Ohta Y, Nakagami G, et al. Compression-induced HIF-1 enhances thrombosis and PAI-1 expression in mouse skin. Wound Repair Regen. 2015;23(5):657-663.
doi pubmed - Jasmiad NB, Abd Ghani RB, Agarwal R, Ismail ZB, Mohd Abdullah AA, Idorus MY. Relationship between serum and tear levels of tissue plasminogen activator and plasminogen activator inhibitor-1 in diabetic retinopathy. BMC Ophthalmol. 2022;22(1):357.
doi pubmed - Yu Y, Li W, Xu L, Wang Y. Circadian rhythm of plasminogen activator inhibitor-1 and cardiovascular complications in type 2 diabetes. Front Endocrinol (Lausanne). 2023;14:1124353.
doi pubmed - Pandey P, Lakhanpal S, Mahmood D, Baldaniya L, Kang HN, Hwang S, Kang S, et al. Recent Update of Natural Compounds as HIF-1alpha Inhibitors in Colorectal Carcinoma. Drug Des Devel Ther. 2025;19:2017-2034.
doi pubmed - Bai M, Xu P, Cheng R, Li N, Cao S, Guo Q, Wang X, et al. ROS-ATM-CHK2 axis stabilizes HIF-1alpha and promotes tumor angiogenesis in hypoxic microenvironment. Oncogene. 2025;44(21):1609-1619.
doi pubmed - Huang C, Tan H, Wang J, Huang L, Liu H, Shi Y, Zhong C, et al. beta-hydroxybutyrate restrains colitis-associated tumorigenesis by inhibiting HIF-1alpha-mediated angiogenesis. Cancer Lett. 2024;593:216940.
doi pubmed - Alam MM, Fermin JM, Knackstedt M, Noonan MJ, Powell T, Goodreau L, Daniel EK, et al. Everolimus downregulates STAT3/HIF-1alpha/VEGF pathway to inhibit angiogenesis and lymphangiogenesis in TP53 mutant head and neck squamous cell carcinoma (HNSCC). Oncotarget. 2023;14:85-95.
doi pubmed - Fujimura T. Significance of PAI-1 on the development of skin cancer: Optimal targets for cancer therapies. Biomed J. 2025;49(1):100850.
doi pubmed - Takayama Y, Hattori N, Hamada H, Masuda T, Omori K, Akita S, Iwamoto H, et al. Inhibition of PAI-1 Limits Tumor Angiogenesis Regardless of Angiogenic Stimuli in Malignant Pleural Mesothelioma. Cancer Res. 2016;76(11):3285-3294.
doi pubmed - Chen Z, Will R, Kim SN, Busch MA, Dunker N, Dammann P, Sure U, et al. Novel function of cancer stem cell marker ALDH1A3 in glioblastoma: pro-angiogenesis through paracrine PAI-1 and IL-8. Cancers (Basel). 2023;15(17):4422.
doi pubmed - Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis, thrombosis and organ dysfunction. Thromb Res. 2012;129(3):290-295.
doi pubmed - Tian M, Liu W, Li X, Zhao P, Shereen MA, Zhu C, Huang S, et al. HIF-1alpha promotes SARS-CoV-2 infection and aggravates inflammatory responses to COVID-19. Signal Transduct Target Ther. 2021;6(1):308.
doi pubmed - Wang JS, Liu X, Xue ZY, Alderman L, Tilan JU, Adenika R, Epstein SE, et al. Effects of aging on time course of neovascularization-related gene expression following acute hindlimb ischemia in mice. Chin Med J (Engl). 2011;124(7):1075-1081.
pubmed - Boe AE, Eren M, Murphy SB, Kamide CE, Ichimura A, Terry D, McAnally D, et al. Plasminogen activator inhibitor-1 antagonist TM5441 attenuates Nomega-nitro-L-arginine methyl ester-induced hypertension and vascular senescence. Circulation. 2013;128(21):2318-2324.
doi pubmed - Salomaa V, Stinson V, Kark JD, Folsom AR, Davis CE, Wu KK. Association of fibrinolytic parameters with early atherosclerosis. The ARIC Study. Atherosclerosis Risk in Communities Study. Circulation. 1995;91(2):284-290.
doi pubmed - Lim W, Park Y, Cho J, Park C, Park J, Park YK, Park H, et al. Estrogen receptor beta inhibits transcriptional activity of hypoxia inducible factor-1 through the downregulation of arylhydrocarbon receptor nuclear translocator. Breast Cancer Res. 2011;13(2):R32.
doi pubmed - Boddy JL, Fox SB, Han C, Campo L, Turley H, Kanga S, Malone PR, et al. The androgen receptor is significantly associated with vascular endothelial growth factor and hypoxia sensing via hypoxia-inducible factors HIF-1a, HIF-2a, and the prolyl hydroxylases in human prostate cancer. Clin Cancer Res. 2005;11(21):7658-7663.
doi pubmed - Mavri A, Alessi MC, Bastelica D, Geel-Georgelin O, Fina F, Sentocnik JT, Stegnar M, et al. Subcutaneous abdominal, but not femoral fat expression of plasminogen activator inhibitor-1 (PAI-1) is related to plasma PAI-1 levels and insulin resistance and decreases after weight loss. Diabetologia. 2001;44(11):2025-2031.
doi pubmed - Roncal C, Orbe J, Rodriguez JA, Belzunce M, Beloqui O, Diez J, Paramo JA. Influence of the 4G/5G PAI-1 genotype on angiotensin II-stimulated human endothelial cells and in patients with hypertension. Cardiovasc Res. 2004;63(1):176-185.
doi pubmed - Cha S, Kim HG, Jang H, Lee J, Chao T, Baek NI, Song IS, et al. Steppogenin suppresses tumor growth and sprouting angiogenesis through inhibition of HIF-1alpha in tumors and DLL4 activity in the endothelium. Phytomedicine. 2023;108:154513.
doi pubmed - Park SY, Jang WJ, Yi EY, Jang JY, Jung Y, Jeong JW, Kim YJ. Melatonin suppresses tumor angiogenesis by inhibiting HIF-1alpha stabilization under hypoxia. J Pineal Res. 2010;48(2):178-184.
doi pubmed - Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781-790.
doi pubmed - Zhu Y, Wang Y, Jia Y, Xu J, Chai Y. Roxadustat promotes angiogenesis through HIF-1alpha/VEGF/VEGFR2 signaling and accelerates cutaneous wound healing in diabetic rats. Wound Repair Regen. 2019;27(4):324-334.
doi pubmed - Yusoff FM, Nakashima A, Kawano KI, Kajikawa M, Kishimoto S, Maruhashi T, Ishiuchi N, et al. Implantation of hypoxia-induced mesenchymal stem cell advances therapeutic angiogenesis. Stem Cells Int. 2022;2022:6795274.
doi pubmed - Voit RA, Sankaran VG. Stabilizing HIF to ameliorate anemia. Cell. 2020;180(1):6.
doi pubmed - Chen MH, Wang YH, Sun BJ, Yu LM, Chen QQ, Han XX, Liu YH. HIF-1alpha activator DMOG inhibits alveolar bone resorption in murine periodontitis by regulating macrophage polarization. Int Immunopharmacol. 2021;99:107901.
doi pubmed - Frischmuth T, Hindberg K, Aukrust P, Ueland T, Braekkan SK, Hansen JB, Morelli VM. Elevated plasma levels of plasminogen activator inhibitor-1 are associated with risk of future incident venous thromboembolism. J Thromb Haemost. 2022;20(7):1618-1626.
doi pubmed - Moideen FM, Rahamathulla MP, Charavu R, Alghofaili F, Sha M, Bhandary YP. PAI-1 influences and curcumin destabilizes MMP-2, MMP-9 and basement membrane proteins during lung injury and fibrosis. Int Immunopharmacol. 2024;143(Pt 3):113587.
doi pubmed - Noguchi R, Kaji K, Namisaki T, Moriya K, Kawaratani H, Kitade M, Takaya H, et al. Novel oral plasminogen activator inhibitor-1 inhibitor TM5275 attenuates hepatic fibrosis under metabolic syndrome via suppression of activated hepatic stellate cells in rats. Mol Med Rep. 2020;22(4):2948-2956.
doi pubmed - Lin LL, Nayak B, Osmulski PA, Wang E, Wang CP, Valente PT, Wang CM, et al. PAI-1 uncouples integrin-beta1 from restrain by membrane-bound beta-catenin to promote collagen fibril remodeling in obesity-related neoplasms. Cell Rep. 2024;43(8):114527.
doi pubmed - Altalhi R, Pechlivani N, Ajjan RA. PAI-1 in diabetes: pathophysiology and role as a therapeutic target. Int J Mol Sci. 2021;22(6):3170.
doi pubmed - Kubala MH, DeClerck YA. The plasminogen activator inhibitor-1 paradox in cancer: a mechanistic understanding. Cancer Metastasis Rev. 2019;38(3):483-492.
doi pubmed - Zhong H, Tang R, Feng JH, Peng YW, Xu QY, Zhou Y, He ZY, et al. Metformin mitigates sepsis-associated pulmonary fibrosis by promoting AMPK activation and inhibiting HIF-1alpha-induced aerobic glycolysis. Shock. 2024;61(2):283-293.
doi pubmed - Sahebkar A, Catena C, Ray KK, Vallejo-Vaz AJ, Reiner Z, Sechi LA, Colussi G. Impact of statin therapy on plasma levels of plasminogen activator inhibitor-1. A systematic review and meta-analysis of randomised controlled trials. Thromb Haemost. 2016;116(1):162-171.
doi pubmed - Kruithof EK. Regulation of plasminogen activator inhibitor type 1 gene expression by inflammatory mediators and statins. Thromb Haemost. 2008;100(6):969-975.
pubmed - Zhou J, Yin G, Yu T, Zhang Y, Tian X, Xia D, Shi L. Rosuvastatin reduces expression of tissue factor through inhibiting RhoA/ROCK pathway to ameliorate atherosclerosis. Panminerva Med. 2021;63(3):402-403.
doi pubmed - Chen J, Shou X, Xu Y, Jin L, Zhu C, Ye X, Mei Z, et al. A network meta-analysis of the efficacy of hypoxia-inducible factor prolyl-hydroxylase inhibitors in dialysis chronic kidney disease. Aging (Albany NY). 2023;15(6):2237-2274.
doi pubmed - Jacoby JJ, Erez B, Korshunova MV, Williams RR, Furutani K, Takahashi O, Kirkpatrick L, et al. Treatment with HIF-1alpha antagonist PX-478 inhibits progression and spread of orthotopic human small cell lung cancer and lung adenocarcinoma in mice. J Thorac Oncol. 2010;5(7):940-949.
doi pubmed - Koh MY, Spivak-Kroizman T, Venturini S, Welsh S, Williams RR, Kirkpatrick DL, Powis G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol Cancer Ther. 2008;7(1):90-100.
doi pubmed - Yang Z, Su W, Wei X, Pan Y, Xing M, Niu L, Feng B, et al. Hypoxia inducible factor-1alpha drives cancer resistance to cuproptosis. Cancer Cell. 2025;43(5):937-954.e939.
doi pubmed - Ilegems E, Bryzgalova G, Correia J, Yesildag B, Berra E, Ruas JL, Pereira TS, et al. HIF-1alpha inhibitor PX-478 preserves pancreatic beta cell function in diabetes. Sci Transl Med. 2022;14(638):eaba9112.
doi pubmed - Yamaoka N, Kodama H, Izuhara Y, Miyata T, Meguro K. Structure-activity relationships of new N-acylanthranilic acid derivatives as plasminogen activator inhibitor-1 inhibitors. Chem Pharm Bull (Tokyo). 2011;59(2):215-224.
doi pubmed - Yasui H, Suzuki Y, Sano H, Suda T, Chida K, Dan T, Miyata T, et al. TM5275 prolongs secreted tissue plasminogen activator retention and enhances fibrinolysis on vascular endothelial cells. Thromb Res. 2013;132(1):100-105.
doi pubmed - Masuda T, Hirata T, Sakamoto T, Tsubata Y, Ichihara E, Kozuki T, Shoda H, et al. Treatment rationale and protocol design: an investigator-initiated phase II study of combination treatment of nivolumab and TM5614, a PAI-1 inhibitor for previously treated patients with non-small cell lung cancer. J Thorac Dis. 2024;16(5):3381-3388.
doi pubmed - Torrente D, Su EJ, Fredriksson L, Warnock M, Bushart D, Mann KM, Emal CD, et al. Compartmentalized actions of the plasminogen activator inhibitors, PAI-1 and Nsp, in ischemic stroke. Transl Stroke Res. 2022;13(5):801-815.
doi pubmed - Shifman SG, O'Connor JL, Radin DP, Sharma A, Infante L, Ferraresso F, Kastrup CJ, et al. Targeting autophagy and plasminogen activator inhibitor-1 increases survival and remodels the tumor microenvironment in glioblastoma. J Exp Clin Cancer Res. 2025;44(1):214.
doi pubmed - Yuan X, Ruan W, Bobrow B, Carmeliet P, Eltzschig HK. Targeting hypoxia-inducible factors: therapeutic opportunities and challenges. Nat Rev Drug Discov. 2024;23(3):175-200.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, including commercial use, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.