| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website https://jocmr.elmerjournals.com |
Review
Volume 17, Number 9, October 2025, pages 469-489

Adenosine Monophosphate-Activated Protein Kinase Activation and Mammalian Target of Rapamycin Complex 1 Inhibition: A Mechanistic Rationale for Anti-Aging Therapy in Type 2 Diabetes
Hidekatsu Yanaia, b, Hiroki Adachia, Mariko Hakoshimaa, Hisayuki Katsuyamaa
aDepartment of Diabetes, Endocrinology and Metabolism, National Kohnodai Medical
Center, Japan Institute for Health Security, Chiba, Japan
bCorresponding Author:
Hidekatsu Yanai, Department of Diabetes, Endocrinology and Metabolism, National Kohnodai Medical
Center, Japan Institute for Health Security, Ichikawa, Chiba 272-8516, Japan
Manuscript submitted August 28, 2025, accepted October 4, 2025, published online October 15,
2025
Short title: AMPK and mTORC1 in Type 2 Diabetes
doi:
https://doi.org/10.14740/jocmr6370
| Abstract | ▴Top |
Aging is a complicated biological process that induces a decline in the human organs’ structure and function and elevates the risks of aging-related diseases such as Alzheimer’s disease (AD) and type 2 diabetes. Type 2 diabetes accelerates all clinical manifestations of aging. Metabolic disorders in type 2 diabetes are unfavorably associated with all hallmarks of aging, such as inflammation and mitochondrial dysfunction. Adenosine monophosphate-activated protein kinase (AMPK) and the mammalian target of rapamycin complex 1 (mTORC1) are key players in cellular metabolism, and AMPK activation and mTORC1 inhibition improve all hallmarks of aging. AMPK activation and mTORC1 inhibition are favorably associated with diabetic complications. Nutritional interventions, such as caloric restriction, resveratrol, and astaxanthin, have AMPK-activating and mTORC1-inhibitory effects and improve metabolic abnormalities in type 2 diabetes. Anti-diabetic drugs, metformin, sodium-glucose cotransporter-2 inhibitors, and glucagon-like peptide 1 receptor agonists have been reported to have AMPK-activating and mTORC1-inhibiting effects and show prevention of aging-related diseases such as cardiovascular disease. The therapeutic interventions that activate AMPK and inhibit mTORC1 may be optimal treatments for type 2 diabetes from the perspective of anti-aging medicine. Furthermore, senolytics may be a promising, direct anti-aging therapeutic strategy specifically for type 2 diabetes and its complications.
Keywords: Aging; AMP-activated protein kinase; Mammalian target of rapamycin complex 1; Metformin; Type 2 diabetes
| Introduction | ▴Top |
Aging is a complicated biological process involving the deterioration of the structure and function of human organs, which increases the risks of aging-related diseases, such as Alzheimer’s disease (AD), cardiovascular diseases (CVDs), and type 2 diabetes [1]. Recent studies have identified several key hallmarks of aging [2, 3]. These include oxidative stress [4], inflammation [5], advanced glycation end products (AGEs) [6], dysbiosis [7, 8], DNA damage [9, 10], telomere shortening [11], epigenetic alterations [12, 13], loss of protein balance [14], deregulated nutrient-sensing [15], altered intercellular communication [16, 17], mitochondrial dysfunction [18, 19], loss of nicotinamide adenine dinucleotide (NAD+) [20, 21], cellular senescence [22], stem cell exhaustion [23, 24], and impaired macroautophagy [25, 26].
Type 2 diabetes [27], aging [28], and related conditions such as frailty [29] and sarcopenia [30] are characterized by chronic inflammation. Hence, reviewing the treatment options and mechanisms between aging and type 2 diabetes is reasonable. Here, we will report the influence of type 2 diabetes on the clinical manifestations and hallmarks of aging. Adenosine monophosphate-activated protein kinase (AMPK) controls intracellular energy balance and regulates cellular metabolism. AMPK signaling declines with aging, enhancing the aging process [31]. AMPK activity is reduced in type 2 diabetes [32]. Therefore, a great number of studies have highlighted that AMPK and its activators may have significant benefits in the treatment of type 2 diabetes. The mammalian target of rapamycin complex 1 (mTORC1) is a key component of cellular metabolism, and inhibition of mTORC1 with therapeutic rapamycin promotes health and longevity in diverse model organisms [33]. Overactivity of mTORC1 has been proposed to provide a new perspective on type 2 diabetes in terms of pathogenesis, clinical focus, and therapeutic strategies [34].
We will provide insights into the effects of AMPK activation and mTORC1 inhibition on hallmarks of aging, type 2 diabetes, and diabetic complications. We searched for the effects of nutritional interventions that have AMPK-activating and mTORC1-inhibitory effects on type 2 diabetes. Furthermore, we investigated whether type 2 diabetes medications, which have been reported to be effective in preventing the onset of complications such as CVD, have AMPK-activating and mTORC1-inhibitory effects. We would like to provide the mechanistic rationale for anti-aging therapy in type 2 diabetes.
| The Influence of Type 2 Diabetes on the Clinical Manifestations of Aging | ▴Top |
The clinical manifestations of aging are shown in Figure 1.
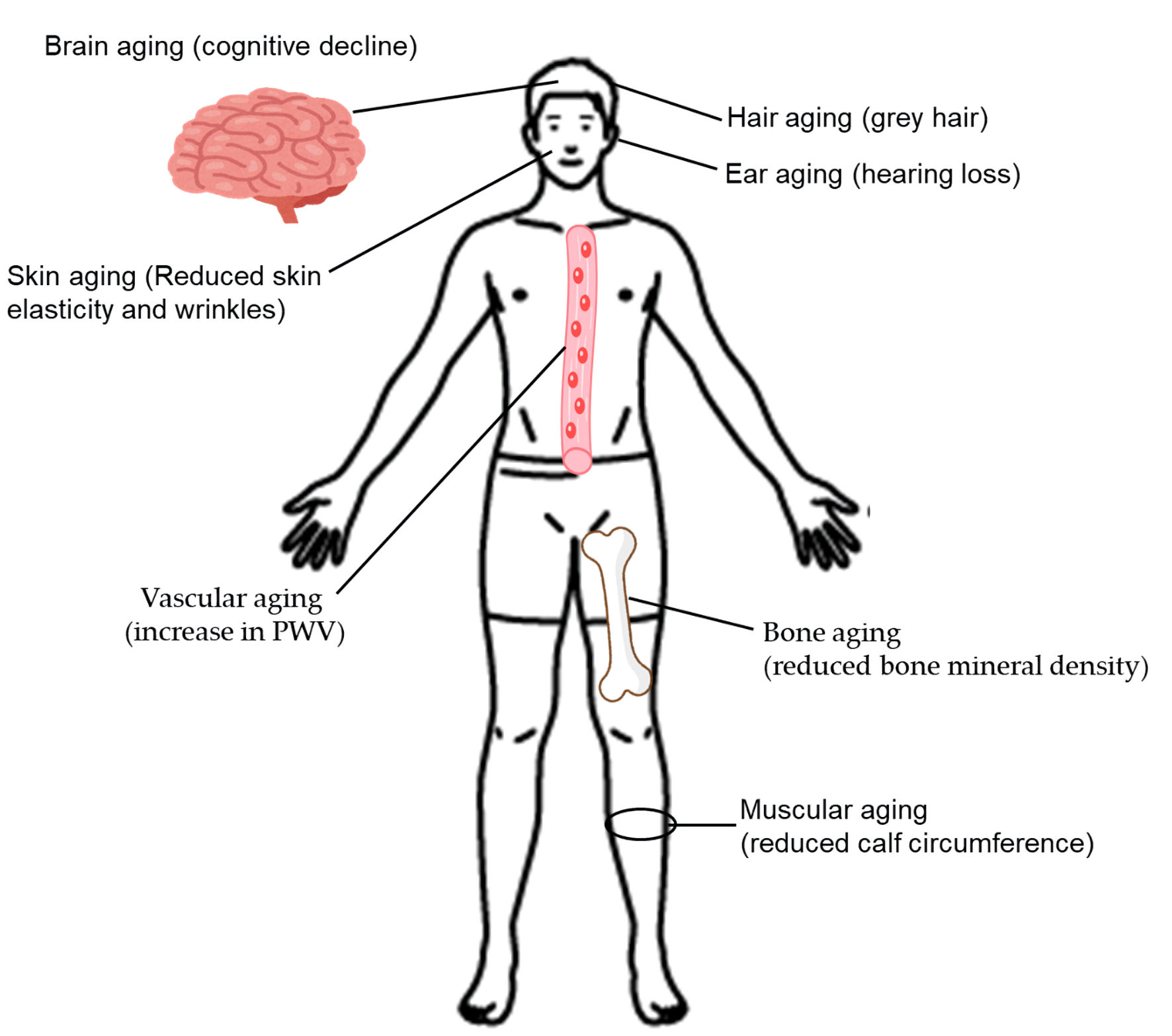 Click for large image |
Figure 1. The clinical manifestations of aging. PWV, pulse wave velocity. |
Hair aging
Dysregulation of the spectral balance of melanocytes in the hair follicle is the fundamental cause of pathological hair greying [35]. As we age, tyrosinase activity decreases, inhibiting melanin synthesis [36]. Premature grey hair is usually accompanied by type 2 diabetes [37].
Skin aging
Diabetes causes increased production of AGEs, which may induce damage to collagen fibers and accelerate skin aging [38]. Diabetic patients had greater epidermal water loss, less elastic skin, and deeper, larger wrinkles [38]. In addition, diabetic patients presented with polycyclic papillae and distorted amorphous collagen fibers [38].
Ear aging
Diabetic patients are twice as likely to experience hearing loss compared to people without diabetes, and people with prediabetes are 30% more likely to experience hearing loss [39].
Brain aging
In a meta-analysis, type 2 diabetic patients were found to have significant impairment in cognition, executive function, and processing speed [40]. Type 2 diabetes was significantly related to the atrophy of gray matter [40].
Bone aging
The meta-analysis revealed that bone mineral density (BMD) was significantly lower in patients with type 2 diabetes than in nondiabetic individuals at the lumbar vertebrae (mean difference (MD): -0.14; 95% confidence interval (CI): -0.22 to -0.06; P = 0.0009), and at femoral neck (MD: -0.11; 95% CI: -0.18 to -0.04; P = 0.002) [41].
Vascular aging
Increased arterial stiffness has been suggested to be a predictor of CVD and all-cause mortality [42-44]. Arterial stiffness increases with age. In postmenopausal women with obesity and diabetes, arterial stiffness is accelerated, leading to increased CVD events compared with lean, non-diabetic premenopausal women [45-47]. The carotid-femoral pulse wave velocity (PWV) is the current standard for assessing arterial stiffness [48, 49]. The meta-analysis showed that PWV was a useful noninvasive early marker for vascular dysfunction in patients with type 2 diabetes [50].
Muscular aging
Insulin resistance is closely associated with increased muscle protein degradation [51]. AGEs accumulation induces skeletal muscle atrophy and dysfunction, and oxidative stress and mitochondrial dysfunction due to type 2 diabetes can induce myocyte apoptosis. Patients with type 2 diabetes are at high risk of developing sarcopenia [52]. Calf circumference measurement was the most effective tool to screen for sarcopenia in patients with type 2 diabetes [53].
Effect of type 2 diabetes on the clinical manifestations of aging
Type 2 diabetes accelerates all clinical manifestations of aging.
| The Hallmarks of Aging and Their Association With Type 2 Diabetes | ▴Top |
Oxidative stress
Oxidative stress is caused by an imbalance between the generation of reactive oxygen species (ROS) and antioxidant capacity and is closely linked to aging and aging-related diseases [54]. Mitochondrial dysfunction due to ROS induces aging [55]. Oxidative damage affects mitochondrial DNA replication and transcription, leading to mitochondrial dysfunction and resulting in increased production of ROS [55]. ROS are highly reactive and can damage cellular components such as DNA, proteins, and lipids, leading to aging [56]. ROS induce mutations and breaks in DNA strands, leading to genomic instability, and DNA damage increases the risk of age-related diseases [57]. Oxidative stress can trigger inflammation and accelerate the shortening of telomeres, which are associated with cellular aging and senescence [58].
Oxidative stress is thought to be involved in the progression of prediabetes to type 2 diabetes [59]. Oxidative stress contributes to insulin resistance, beta-cell dysfunction, and diabetic complications. The levels of glutathione, a major antioxidant, are notably low in patients with type 2 diabetes, exacerbating oxidative stress and inflammation [60]. Elevated interleukin-6 (IL-6) levels further intensify inflammation and oxidative stress, disrupt insulin signaling, and exacerbate diabetic complications. Patients with type 2 diabetes are prone to inflammation and oxidative stress [61, 62], and hyperglycemia, hyperglycemia-induced oxidative stress and inflammation are closely related to the development and progression of type 2 diabetes and its complications.
Inflammation
Aging is associated with increased blood levels of inflammatory markers, a condition termed “inflammaging” [63]. The inflammaging contributes to many aging-related pathologies. Inflammation is a major cause of type 2 diabetes and CVD [64]. Inflammatory markers predict the development of type 2 diabetes and diabetic complications. Overnutrition, unhealthy diets, lack of exercise, obesity, and aging are all known to increase inflammation, promote insulin resistance, and perpetuate the development of type 2 diabetes. The proinflammatory cells infiltration into adipose tissue is associated with an increased production of inflammatory chemokines and a reduced adiponectin [65]. Inflammation in adipose tissue induces insulin resistance and type 2 diabetes in obesity. In addition to adipose tissue, liver, muscle, and pancreas are the sites of inflammation in the presence of obesity [66], inducing insulin resistance and beta-cell dysfunction. Furthermore, the nuclear factor kappa B (NF-κB) and the nod-like receptor pyrin 3 (NLRP3) inflammasome axis leads to pancreatic beta-cell dysfunction and the development of type 2 diabetes [67].
AGEs
AGEs are formed by non-enzymatic glycation of proteins and lipids [68]. AGEs accumulation in tissues is significantly correlated with blood glucose levels [69]. The accumulation of AGEs in nucleotides, lipids, and proteins is a crucial factor in aging [70]. AGEs are associated with changes seen during the aging process and the development of age-related diseases. Even after an improvement in hyperglycemia, AGE levels in diabetic tissues often do not return to normal, which may indicate “metabolic memory” [71]. AGEs are implicated in the development of diabetes [72]. AGEs bind to the receptor for AGEs (RAGE), thereby activating proinflammatory signaling pathways [73]. AGEs bind RAGE, triggering multiple intracellular signaling pathways, including the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase that leads to increased production of ROS, thus contributing to generating a pro-oxidant environment [74], contributing to the development of diabetic microvascular complications [75].
Dysbiosis
The gut microbiome plays a key role in regulating health and disease. With aging, profound changes in the composition and function of the gut microbiome can lead to gut dysbiosis, increased inflammation, weakened immunity, and metabolic disorders [76]. Inflammaging is significantly associated with gut microbial alterations and contributes to the development and progression of AD, CVD, obesity, and diabetes [76]. A systematic review has reported that a major change in the gut microbiota in obesity is characterized by a specific decrease in butyrate-producing bacteria and the production of metabolites and components that lead to insulin resistance, type 2 diabetes, and CVD [77]. Inflammation plays a major role as a link between gut microbiota dysbiosis and obesity-associated metabolic complications. Dysbiosis may contribute to the development of type 2 diabetes [78]. Individuals with type 2 diabetes exhibit a notable reduction in butyrate-producing bacteria [78]. Gut dysbiosis can cause inflammation and induce peripheral and brain insulin resistance, potentially amplifying processes that promote the progression of AD. Furthermore, gut dysbiosis has been shown to increase the risk of developing AD [79].
DNA damage
The accumulation of DNA damage contributes to cell death, senescence, and tissue dysfunction [9]. DNA damage induces inflammation through the activation of the interferon gene axis and NF-κB [9]. Loss of efficient repair of DNA can lead to accelerated aging [10]. High levels of mutations in DNA induce excessive cell death and stem cell exhaustion [10]. Accumulated DNA damage and mutation are the hallmarks of aging. DNA damage induces a senescence-associated secretory phenotype (SASP), such as increased secretion of inflammatory cytokines [80]. SASP may also play important roles in diabetes. White adipose tissue (WAT) cells are prone to senescence due to obesity and type 2 diabetes. The senescence of WAT induces hypertrophy of adipocytes, insulin resistance, and dyslipidemia [80]. The meta-analysis showed that significantly elevated rates of chromosomal aberrations have been observed in type 2 diabetic subjects [81].
Telomere shortening
Telomere is a part of a chromosome that prevents the tangling of the chromosome during DNA replication. When a cell divides, the telomere gets shorter each time, and this process continues until the telomere becomes too short, at which point the cell cannot divide anymore, and the cell dies [82]. Oxidative stress and incomplete DNA replication play a major role in telomere shortening [83]. When telomeres shorten and reach a critical length, apoptosis is initiated, which contributes to cellular senescence [84].
In patients with diabetes, excessive oxidative stress damages telomeres, shortening their length [85]. Telomere length is a good surrogate marker for mortality and diabetic complications in diabetic patients. Telomere length in pancreatic beta-cells is also shortened in diabetic patients, which induces impaired proliferation and insulin secretion [85]. Telomere attrition in adipose tissue induces insulin resistance. Excessive oxidative DNA damage and telomere shortening in type 2 diabetes have been suggested to lead to senescent retinal, renal, and vascular phenotypes [86].
Epigenetic alterations
Among epigenetic alterations, DNA methylation, the involvement of noncoding RNAs, and histone modifications play important roles in aging [87]. Aberrant DNA methylation influences cellular functions and may lead to CVD, metabolic diseases, and cancer. Long noncoding RNAs are increasingly recognized as key regulators in cellular senescence and various age-related pathologies [88]. In general, histones are decreased, and aberrant nucleosome occupancies occur during aging. The accumulation of histone variants is another common feature in the aging process [89]. Methylation and acetylation of histone play an important role in regulating the chromatin structure [90], and such changes influence the senescence-associated gene expression [91]. Unhealthy eating habits and lack of exercise increase the risk of obesity and can affect gene expression through epigenetic alterations [92]. Epigenetic alterations occur early in obesity and may contribute to obesity-related complications [92]. Patients with type 2 diabetes exhibit epigenetic alterations associated with mitochondrial dysfunction in pancreatic islets [93]. DNA methylation, histone modifications, and regulation by noncoding RNAs are epigenetic alterations observed in type 2 diabetes [94]. Oxidative stress can induce abnormal DNA methylation in beta cells, leading to altered gene expression related to insulin secretion [95]. In type 2 diabetes, hyperglycemia induces DNA methyltransferase 1-dependent epigenetic reprogramming of the endothelial exosome proteome [96]. In middle-aged and older Australians, epigenetic age was positively associated with the prevalence and incidence of type 2 diabetes [97].
Loss of protein balance
Aging is associated with a decline in muscle mass, strength, and function, a process known as sarcopenia, which is largely attributed to an imbalance between muscle protein synthesis and breakdown [98]. This imbalance is influenced by factors like reduced anabolic response to stimuli like food intake and exercise, as well as changes in protein absorption. The muscle protein synthetic response to the major anabolic stimuli, such as food intake and exercise, tends to be blunted in elderly people [99-101]. This anabolic resistance is now considered to be a key factor contributing to progressive loss of skeletal muscle mass. Diabetes can lead to pronounced physiological and cellular adaptations in protein regulation [102], and such pathological conditions can result in a net loss of muscle mass [103]. Muscle strength is reduced in individuals with insulin resistance and type 2 diabetes [104, 105].
Deregulated nutrient sensing
Deregulated nutrient sensing is a key hallmark of aging, where cellular and organismal processes become less effective at responding to nutrient availability. This dysregulation, particularly in pathways like mTORC1 signaling, contributes to the aging process [106]. The nutrient-sensing network (NSN) is an interconnected network of signaling pathways centered on insulin, insulin-like growth factor-1 (IGF-1), mTORC1, AMPK, and sirtuins (SIRTs), the disruption of which promotes aging [3]. Lifestyle factors such as diet may modulate the NSN [107, 108].
Altered intercellular communication
Age-related changes in intercellular communication include SASP, direct intercellular communication via gap junctions or tubular structures, and long-distance communication via extracellular vesicles, and paracrine communication via hemichannels, including connexins [109]. Extracellular vesicles (EVs) are lipid membranes that are produced and released from parent cells and taken up by recipient cells [110]. EV-mediated communication between adipocytes, blood vessels, and immune cells may play an important role in the development of type 2 diabetes [110]. Impaired Ca2+ uptake into mitochondria, or impaired interconnected mitochondrial network, is associated with defective insulin secretion [111]. Altered mitochondrial metabolism may also impair beta-cell-beta-cell communication.
Mitochondrial dysfunction
Mitochondrial dysfunction is a hallmark of aging, significantly impacting cellular energy, oxidative balance, and calcium levels [112]. Mitochondrial dysfunction can trigger cellular senescence [112]. Hyperglycemia, hyperlipidemia, and insulin resistance contribute to mitochondrial dysfunction in every organ system [113]. Mitochondria are at the crossroads of important intracellular pathways, such as energy substrate metabolism, ROS production, and apoptosis. Mitochondrial dysfunction induces impaired oxidative phosphorylation, increases ROS generation, mitochondrial DNA damage, and altered mitochondrial dynamics, which contribute to the development of diabetic complications [114]. Mitochondrial dysfunction induces dysfunction of the liver, heart, skeletal muscle, beta-cells, and nervous system in type 2 diabetes [113]. Furthermore, the accumulation of mitochondrial DNA mutations and mitochondrial DNA copy number depletion, as well as epigenetic modification of the mitochondrial genome, are associated with the development of type 2 diabetes.
A decline in NAD+
NAD+ is a crucial coenzyme involved in numerous cellular processes, including energy production and DNA repair. Most of the cellular NAD+ is generated through nicotinamide phosphoribosyl transferase (NAMPT) activation, a key rate-limiting enzyme that is involved in the salvage pathway. NAD+ is essential for mitochondrial function, and NAD+ acts as a cofactor for enzymes involved in the electron transport chain [115]. Furthermore, NAD+ is known to benefit and restore DNA replication, chromatin, epigenetic modifications, and gene expression. A decline in NAD+ is strongly linked to the aging process and the development of aging-related diseases [116]. As we age, NAD+ levels naturally decrease, potentially impairing these vital cellular functions and contributing to aging-related decline and disease [117]. NAD+ is depleted in diabetes, CVD, and neurodegenerative diseases, and in aging [118]. An important hallmark of dysfunctional obese adipose tissue is impaired NAD+/SIRT signaling [119].
Cellular senescence
Cellular senescence, a state of irreversible cell cycle arrest, is a key hallmark of aging. It is a cellular response to stress and damage that can contribute to age-related diseases and the overall aging process [120]. Cell senescence accumulation with age can be detrimental, driving chronic inflammation and tissue dysfunction [121]. Senescence of islet beta-cells induces the pathogenesis of type 2 diabetes. Senescence-associated beta-galactosidase activity is increased in pancreatic islet beta cells isolated from aged and type 2 diabetic mice [122]. Furthermore, the expression level of IGF-1 receptor (IGF-1R) in beta cells of type 2 diabetes model mice was higher than that of control mice, suggesting a significant association between beta-cell senescence and type 2 diabetes [123]. Aging pancreatic islet beta cells have a reduced insulin secretory capacity and are less sensitive to high glucose stimulation [124]. A high-calorie diet can promote adipocyte senescence and cause insulin resistance [125], and insulin resistance can further induce pancreatic beta-cell senescence [126].
Stem cell exhaustion
Stem cell exhaustion is a significant factor in aging, characterized by a decline in the ability of stem cells to regenerate and maintain tissue homeostasis [127]. Patients with type 2 diabetes may have a reduced ability to revascularize ischemic tissues due to abnormal production of circulating pro-vascular progenitor cells [128]. Increasing oxidative stress and inflammation intensify this regenerative cell exhaustion process. Diabetes reduces circulating stem/progenitor cells and impairs their function [129]. Diabetes inhibits bone marrow stem/progenitor cell mobilization, adversely affecting physiological hematopoiesis, immunoregulation, and tissue regeneration.
Impaired macroautophagy
Autophagy is a fundamental cellular process that removes molecules and intracellular elements, such as nucleic acids, proteins, lipids, and organelles, through lysosome-mediated degradation, promoting homeostasis, differentiation, development, and survival [130]. Although autophagy declines with age, it is a key determinant of cellular health and organismal longevity, and impaired or imbalanced autophagy promotes pathological aging and disease [130]. The decline in macroautophagy with age has been implicated in aging-related diseases such as Parkinson’s disease and AD, cancer, and CVD [131]. Autophagy-deficient mice exhibit hypoinsulinemia and hyperglycemia, suggesting that autophagy is required for maintaining pancreatic beta-cell structure, mass, and function [132]. In diabetes, the accumulation of AGEs, excess oxidative stress, endoplasmic reticulum stress, hypoxia, and the activation of the renin-angiotensin system regulate autophagy activity, which contributes to the development of diabetic nephropathy [133]. Enhanced macroautophagy is important for maintaining the fidelity of the secretory apparatus under conditions of high insulin demand, and failure of these adaptive mechanisms likely contributes to beta-cell dysfunction [134]. Islet macroautophagic impairment was found in human type 1 diabetes [135]. Endothelial dysfunction is a key determinant in the pathogenesis of diabetes. Autophagy appears to play an important role in endothelial cells, ensuring endothelial homeostasis and functions. Autophagic flux impairment has been observed in patients with type 1 or type 2 diabetes [136]. Diabetes causes impaired autophagy in endothelial cells, contributing to diabetes-mediated endothelial dysfunction.
The association between hallmarks of aging and type 2 diabetes
Metabolic disorders such as hyperglycemia and insulin resistance observed in type 2 diabetes induce harmful factors such as oxidative stress, inflammation, AGEs, and dysbiosis. Such harmful factors induce impaired maintenance of healthy cells by inducing DNA damage, epigenetic alteration, mitochondrial dysfunction, worsening type 2 diabetes, and developing diabetic complications. Impaired maintenance of healthy cells is induced by an impaired clearance of senescent cells due to stem cell exhaustion and impaired macroautophagy. Impaired clearance of senescent cells is unfavorably associated with metabolic disorders and diabetic complications. Thus, type 2 diabetes and aging form a vicious cycle, exacerbating each other’s pathology.
| The Significance of AMPK Activation and mTORC1 Inhibition for Anti-Aging Medicine | ▴Top |
AMPK activation is strongly linked to anti-aging mechanisms, as it acts as a cellular energy sensor and regulator that maintains cellular homeostasis, metabolism, stress resistance, and promotes processes like autophagy, all of which are crucial for healthy aging and extended lifespan [137]. mTORC1 and AMPK signaling play a crucial role in biological networks involved in cellular senescence [138]. AMPK activation and mTORC1 inhibition are closely linked mechanisms involved in the aging process and are expected to have the effect of extending lifespan and reducing diseases associated with aging. AMPK is activated under low ATP levels, leading to the inhibition of mTORC1, a key pathway involved in cell growth and metabolism that is often dysregulated with age [138].
In acute energy-deficient status, AMPK activation is protective to restore cellular energy by stimulating catabolism and inhibiting anabolism [139]. However, in chronic conditions, overactivation of AMPK can be detrimental. Chronic activation has been proposed to inhibit overall energy metabolism by shifting processes toward energy-dissipating states in the heart [140].
mTORC1 activation induces muscle growth (hypertrophy) by increasing protein synthesis [141]. Complete and chronic inhibition can lead to muscle atrophy and dysfunction [141]. Chronic mTOR inhibition by rapamycin induces muscle insulin resistance [142]. mTORC1 activity is important for immune cell function, and pharmacological inhibition of mTORC1 can significantly alter or eliminate immune responses [143]. Over-inhibition can impair lipid metabolism, mitochondrial function, and autophagy [144, 145]. mTOR signaling is necessary for rapid wound healing, and mTOR signaling enables an increase in cell volume that facilitates wound repair [146]. Therefore, restoring balance and regulation of mTORC1 is key to anti-aging.
Effects of AMPK activation and mTOR inhibition on aging-related molecules and aging hallmarks were shown in Figure 2. Nuclear factor E2-related factor 2 (Nrf2) is activated under oxidative stress conditions, which promotes antioxidant gene expression [147]. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α) and Nrf2 have been shown to promote each other and regulate the expression of antioxidant genes. Nrf2 is a downstream target of AMPK, and activation of the AMPK/Nrf2 signaling pathway is important for preventing aging-related diseases.
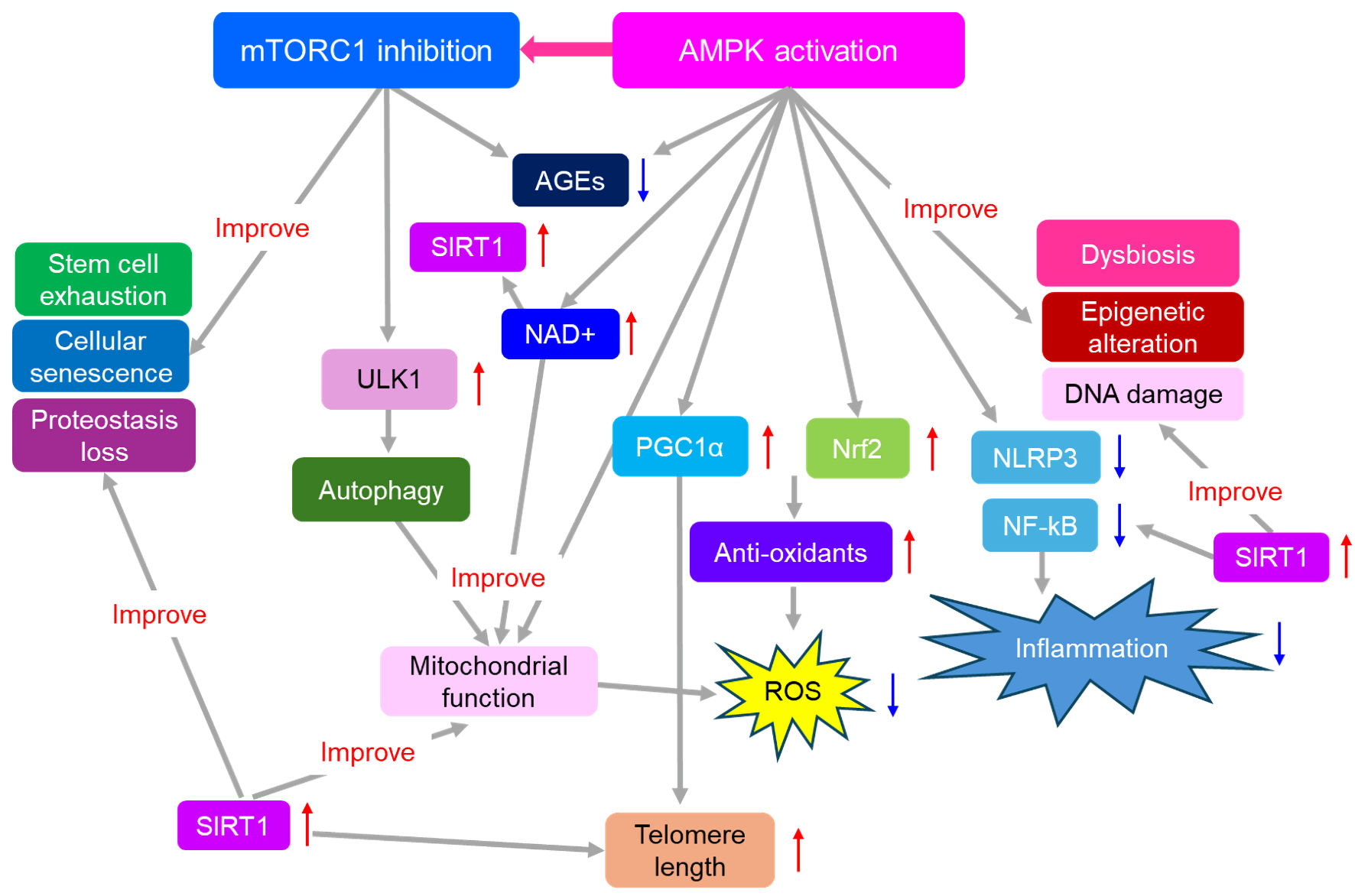 Click for large image |
Figure 2. Effects of AMPK activation and mTOR inhibition on aging-related molecules and aging hallmarks. Thick gray arrows indicate effects, and red upward arrows indicate the increase, and blue downward arrows indicate the decrease. AGEs: advanced glycation end products; AMPK: adenosine monophosphate-activated protein kinase; mTORC1: mammalian target of rapamycin complex 1; NAD+: nicotinamide adenine dinucleotide; NLRP3: nod-like receptor pyrin 3 inflammasome; Nrf2: nuclear factor E2-related factor 2; NF-κB: nuclear factor-kappa B; PGC1α: peroxisome proliferator-activated receptor-γ coactivator 1α; ROS: reactive oxygen species; SIRT1: sirtuin 1; ULK1: unc-51-like autophagy activating kinase 1. |
Chronic inflammation associated with increased NF-κB signaling is a typical feature of several age-related metabolic disorders [148]. Activators of AMPK have anti-inflammatory properties. AMPK activation can inhibit NF-κB signaling [149]. AMPK activation can also inhibit the NLRP3 inflammasome pathway [150].
AMPK activation mitigates AGEs by reducing inflammation and cellular damage [70]. The inhibition of mTORC1 has been suggested to diminish the accumulation of AGEs, possibly due to a protective role of autophagy in the clearance of AGEs [151]. AMPK inactivation is an etiological factor in intestinal dysfunction, and AMPK activation favorably influences intestinal health [152].
AMPK acts as a central regulator of epigenetic processes by phosphorylating histones, DNA methyltransferases, and histone modifiers [153]. Epigenetic modifications play an important role in the management of DNA damage, influencing both the detection and repair of DNA damage and leading to long-term alterations in gene expression [154]. Telomerase reverse transcriptase (TERT) has a telomere-lengthening effect [155]. AMPK-dependent PGC1α upregulation is required for the metformin-induced upregulation of TERT and telomere activity and length [156].
AMPK controls autophagy through mTORC1 and unc-51-like autophagy activating kinase 1 (ULK1) signaling, which augments the quality of cellular housekeeping. The ULK1 complex plays a central role in the initiation stage of autophagy [157]. mTORC1 inhibits the activity of the ULK1 complex by phosphorylating ULK1 and Atg13 [158].
AMPK is promptly activated after mitochondrial stress [159]. AMPK regulates autophagy and mitophagy by activating the kinase ULK1 [160]. AMPK phosphorylates mitochondrial fission factors to promote mitochondrial fission under energy stress. By simultaneously regulating mitochondrial fission, mitophagy, and the transcriptional regulation of mitochondrial biogenesis, AMPK functions as a signal integration platform to maintain mitochondrial function.
NAD+ is a vital coenzyme crucial for maintaining mitochondrial function, directly participating in key energy-producing pathways like the tricarboxylic acid cycle and oxidative phosphorylation, and playing a role in cellular signaling and overall homeostasis [161]. Declining NAD+ levels are associated with aging and various chronic diseases, often accompanied by mitochondrial dysfunction [161]. AMPK cooperates with another metabolic sensor, the NAD+-dependent type III deacetylase SIRT1, to regulate the expression of genes involved in energy metabolism [162]. AMPK increases the activity of SIRT1 by increasing intracellular NAD+ levels.
The anti-aging effects of SIRT1 have been widely studied [163, 164]. SIRT1 regulates inflammation and stress resistance and suppresses various effects associated with aging, including genomic instability, stem cell depletion, mitochondrial dysfunction, and telomere shortening. Activating SIRT1 inhibits inflammation by deacetylating NF-κB [165]. AMPK activation and mTORC1 inhibition improve all hallmarks of aging.
| Effects of AMPK Activation and mTORC1 Inhibition on Diabetic Complications | ▴Top |
Diabetic complications include microangiopathy, such as neuropathy, retinopathy, and nephropathy, and macroangiopathy, such as CVD. Research is increasingly highlighting the link between AD and type 2 diabetes, showing that the risk of developing both increases exponentially with age, and that type 2 diabetes predisposes to AD [166]. What about the effects of AMPK activation and mTOR inhibition on these diabetic complications and AD?
Diabetic neuropathy
Increased ROS, amplified apoptosis, decreased insulin secretion, neuroinflammation, and decreased autophagy induce the development of diabetic neuropathy [167]. Impaired AMPK signaling in neurons is associated with mitochondrial dysfunction and peripheral neuropathy in diabetic models [168]. Activation of AMPK has been shown to improve heat sensitivity and reduce epidermal innervation density in streptozotocin-treated rodents [168].
Tang Bi formula (TBF) has been proven effective for diabetic polyneuropathy. Very recently, TBF ameliorated diabetic polyneuropathy by rectifying mitochondrial dynamic imbalance and modulating the activation of the AMPK-PGC-1α-mitochondrial fusion protein pathway [169].
The altered mTORC1 activity can impair nerve function by affecting brain-derived neurotrophic factor (BDNF) in Schwann cells, increasing ROS and AGEs, and suppressing autophagy [170]. Targeting the mTOR pathway with inhibitors or natural compounds shows promise for novel regenerative therapeutic strategies against diabetic complications like neuropathy [171].
Diabetic retinopathy (DR)
C1q/tumor necrosis factor-related protein-3 (CTRP3), a newly discovered adipokine, prevents diabetes-induced retinal vascular permeability by activating AMPK [172]. Recently, mTORC1 inhibition has been reported as a novel gene therapeutic strategy for DR [173].
Diabetic nephropathy
Renal tubulointerstitial fibrosis is a key pathological feature of diabetic nephropathy, and renal tubular damage may be associated with abnormal mitophagy. The AMPK agonist improved renal oxidative stress and tubulointerstitial fibrosis in high-fat diet- and streptozotocin-induced type 2 diabetic mice by activating the AMPK signaling pathway [174].
It has been observed that mTORC1 is involved in the progression of diabetic nephropathy by inhibiting autophagy, promoting inflammation, and increasing oxidative stress [175]. The inhibitory effect of rapamycin on the development and progression of diabetic kidney disease (DKD) was observed in diabetic animal models [176].
CVD
Emerging evidence suggests that naturally occurring AMPK activators may have significant cardiovascular (CV) benefits [177]. AMPK activators prevent CVD by reducing blood pressure, plasma glucose, serum lipids, and ROS production, and by increasing NO bioavailability.
The mTORC1 pathway plays a crucial role in CVD, promoting processes like atherosclerosis, myocardial infarction, and heart failure through effects on cell growth, metabolism, and inflammation [178]. Inhibiting mTORC1 can offer cardioprotective effects, reducing infarct size and slowing disease progression [179].
AD
AMPK reduces tau phosphorylation and improves brain function in an AD-like model [180]. Aberrant mTORC1 signaling is associated with AD pathogenesis, contributing to hallmark pathologies like amyloid beta plaques and tau neurofibrillary tangles through increased production and decreased clearance [181, 182]. mTORC1 activation also promotes neuroinflammation, oxidative stress, and cognitive decline, while its inhibition, particularly with drugs like rapamycin, has shown promise in preclinical models for clearing amyloid and tau aggregates and improving cognitive function [181, 182].
| Sex Differences in the Anti-Aging Medicine | ▴Top |
The gradual decline in hormone production and action with aging has detrimental effects on human health, including an increased risk of chronic diseases and a shortened lifespan [183]. Hormonal changes contribute to a variety of chronic diseases, including frailty, diabetes, CVD, and dementia.
Osteoarthritis (OA) affects women more frequently than men, especially after menopause [184, 185]. Cartilage undergoes various changes with aging, including a decrease in cell density and extracellular matrix content, followed by thinning and fragility [186]. The significant increase in the prevalence of OA in postmenopausal women suggests a link between OA and a decrease in estrogen [184].
The biological actions of estrogen are mediated by the estrogen receptor α or β (ERα or ERβ), which are members of a broad nuclear receptor superfamily. The reduction in circulating estrogen, regulated by classical ERα and ERβ, led to rapid changes in pancreatic beta-cell and islet function, glucose transporter 4 (GLUT4) expression, insulin sensitivity and glucose tolerance, lipid homeostasis dysfunction, oxidative stress, and inflammatory cascades [187]. Clinical trials have shown that hormone replacement therapy has a protective effect on glucose metabolism and may be useful for treating type 2 diabetes in menopausal women [187].
Early menopause and premature ovarian failure are independent risk factors for type 2 diabetes [188]. Patients with type 1 diabetes enter menopause earlier than controls [189]. Follicular development and growth are adversely affected by chronic hyperglycemia, which may lead to ovarian failure in type 1 diabetes. AGEs may contribute to ovarian aging [190].
Testosterone levels in men begin to decline between the ages of 30 and 40 and continue to decline until death [191]. Local and systemic illness, prescription medications, lung tumors, excessive smoking or alcohol, obesity, and untreated diabetes were correlated with a decrease in testosterone [192]. Testosterone levels have been reported in untreated elderly diabetic males to be approximately 15% lower compared to their non-diabetic counterparts [193]. Low testosterone levels are closely associated with visceral obesity, insulin resistance, poor glycemic control, and prolonged diabetes duration [194]. Low testosterone levels are associated with CVD, metabolic syndrome, insulin resistance, type 2 diabetes [192], psychological status [191], reduced muscle and bone mass, and sexual dysfunction [195].
| Effects of Exercise on AMPK Activation, mTORC1 Inhibition, Type 2 Diabetes, and Aging-Related Diseases | ▴Top |
During exercise, AMPK is activated in skeletal muscles in humans, and exercise may be the most powerful physiological activator of AMPK [196]. mTORC1 is inhibited in liver and fat by exercise, which may underlie some of the health benefits in these tissues [197].
A recent meta-analysis showed that combined resistance and aerobic exercise intervention significantly improved fasting blood glucose and serum lipids in patients with type 2 diabetes, especially in terms of blood glucose control and CV risk, demonstrating better outcomes than aerobic exercise alone [198].
The meta-analysis provides strong evidence that structured exercise-based programs significantly reduce the risk of major adverse cardiovascular events (MACE) in patients with CVD [199].
A meta-analysis showed that the Mini-Mental State Examination (MMSE) scores of the exercise group were higher than those of the control group (MD: 2.24; P = 0.002) [200]. Low-intensity aerobic exercise improved cognitive and motor function in patients with AD, while strength training and high-intensity exercise had little effect. This may be related to AMPK activation and mTORC1 inhibition caused by exercise intensity.
| Effects of Nutritional Interventions That Have AMPK-Activating and mTORC1-Inhibitory Effects on Type 2 Diabetes | ▴Top |
Effects of nutritional interventions that have AMPK-activating and mTORC1-inhibitory effects on type 2 diabetes are shown in Table 1 [201-217].
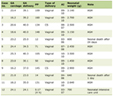 Click to view |
Table 1. Effects of Nutritional Interventions
That Improve Aging Hallmarks by AMPK Activation and mTORC1 Inhibition on Type 2
Diabetes |
Calorie restriction has both AMPK-activating and mTORC1-inhibitory effects [201, 202]. At 6 months, each 500-kcal/day decrease in energy intake resulted in clinically meaningful reductions in body weight (MD: -6.33 kg; 95% CI: -7.76 to -4.9) and glycated hemoglobin (HbA1c, MD: -0.82%; 95% CI: -1.05 to -0.59) [203]. However, caloric restriction can be harmful for frail elderly individuals, increasing the risk of sarcopenia, functional decline, and weakened immune function. Frail older adults have a lower threshold for functional impairment, and weight loss from calorie restriction can lead to significant risks and lower quality of life [218]. Calorie restriction can induce diabetic ketoacidosis, which is life-threatening, in patients with poorly controlled diabetes [219].
Vitamin E shows AMPK-activating and mTORC1-inhibitory effects [204, 205]. In studies with an intervention period of less than 10 weeks, vitamin E supplementation significantly reduced fasting blood glucose levels [206]. Nonlinear dose-response analysis showed that the most effective dose range of vitamin E for lowering HbA1c in diabetic patients was 400 - 1,300 mg/day, with the highest effect observed at a dose of 1,000 mg/day [206]. Vitamin C also has AMPK-activating and mTORC1-inhibitory effects [208,208]. The long-term (≥ 12 weeks) and high-dose vitamin C supplementation (≥ 1,000 mg/day) may ameliorate glycemic profile in type 2 diabetic patients [209]. However, additional high-quality randomized controlled trials (RCTs) are necessary to validate these results.
Resveratrol has both AMPK-activating and mTORC1-inhibitory effects [210, 211]. A meta-analysis of 17 RCTs in 871 patients with type 2 diabetes found that ≥ 500 mg of resveratrol was superior to placebo in improving fasting plasma glucose (MD: -13.34 mg/dL; 95% CI: -22.73 to -3.95, P = 0.005) and HbA1c (MD: -0.41%; 95% CI: -0.65 to -0.16, P = 0.001) at 3 months [212]. Astaxanthin has both AMPK-activating and mTORC1-inhibitory effects [213]. After 12 weeks of supplementation with 12 mg of astaxanthin, blood glucose levels at 120 min after a 75 g oral glucose tolerance test were significantly reduced compared to baseline; furthermore, HbA1c levels decreased from 5.64±0.33% to 5.57±0.39% (P < 0.05) [214]. The Matsuda index, an indicator of insulin resistance, also improved in the astaxanthin group compared to baseline [214]. The n3-polyunsaturated fatty acids (PUFA) have been reported to have both AMPK-activating and mTORC1-inhibitory effects [215, 216]. In an individual participant-level pooling project of 20 prospective cohort studies, a total of 16,693 incident type 2 diabetes cases were identified during follow-up (median follow-up ranging from 2.5 to 21.2 years). In pooled multivariable analysis, eicosapentaenoic acid (EPA) (-8%) and docosahexaenoic acid (DHA) (-18%) were associated with a lower incidence of type 2 diabetes [217]. In a population pharmacokinetic-pharmacodynamic modelling and intake threshold study, a daily intake of n3-PUFA at 0.4 g was sufficient to achieve an HbA1c level of 7% in more than 95% of patients [220].
However, we should mention that the evidence for many of these (resveratrol, astaxanthin) in human diabetes is often from small studies, and meta-analyses show mixed or minimal clinical effects compared to pharmaceuticals. We should acknowledge the limitations of the human evidence for these supplements, including bioavailability issues and the magnitude of effect compared to standard pharmacotherapy.
| Effects of Anti-Diabetic Drugs That Have AMPK-Activating and mTORC1-Inhibitory Effects on Aging-Related Diseases | ▴Top |
Effects of anti-diabetic drugs that have AMPK-activating and mTORC1-inhibitory effects on aging-related diseases are shown in Table 2 [221-230].
Click to view |
Table 2. Effects of Anti-Diabetic Drugs That
Have AMPK-Activating and mTORC1-Inhibitory Effects on Aging-Related
Diseases |
Metformin has been reported to have both AMPK-activating and mTORC1-inhibitory effects [231, 232]. The UK Prospective Diabetes Study (UKPDS) demonstrated a legacy effect of early, intensive metformin therapy for type 2 diabetes in overweight patients, where the reduction in myocardial infarction and death from any cause persisted even after the trial concluded and glycemic differences were lost [233]. This “legacy effect” showed that early and thorough treatment of type 2 diabetes is crucial, as a period of intensive blood glucose control with metformin provided long-term health benefits that continued to show up years later. AMPK activation by metformin may counteract detrimental metabolic memory by restoring cellular energy balance, improving insulin sensitivity, and reducing fibrosis and inflammation, which are hallmarks of metabolic memory [234]. Such a mechanism restores cellular function, mitigates age-related damage, and prevents the establishment of aging-related diseases such as CVD and AD.
A recent umbrella review of systematic reviews with meta-analysis showed that the risk of CVD mortality in metformin patients was lower than in the other two groups (placebo and other anti-diabetic drugs) (odds ratio (OR): 0.771; 95% CI: 0.688 to 0.853), and the risk of CVD in metformin users was also lower than in the other two groups (OR: 0.828; 95% CI: 0.781 to 0.785) [221]. A meta-analysis of 10 clinical observational studies showed no significant association between AD incidence and metformin exposure (OR: 1.17; 95% CI: 0.88 to 1.56; P = 0.291) [222]. Further well-designed RCTs are required to understand the significance of metformin in the prevention of AD. Three clinical trials, Metformin in Alzheimer’s Dementia Prevention (MAP), MET-FINGER, MET-MEMORY, are currently underway to understand the effect of metformin on the prevention of AD [223].
Sodium-glucose cotransporter-2 inhibitors (SGLT2is) have both AMPK-activating and mTORC1-inhibitory effects [235, 236]. The EMPA-REG OUTCOME demonstrated that empagliflozin reduced the incidence of the 3-point MACE (death from CV causes, nonfatal myocardial infarction, or nonfatal stroke) by 14% as compared with placebo [224]. The CANVAS program also demonstrated that the incidence of the 3-point MACE was lower with canagliflozin as compared with placebo by 14% [225]. A retrospective cohort study including 1,348,362 patients with type 2 diabetes showed that SGLT2i use was associated with a lower risk of AD (adjusted hazard ratio (aHR): 0.81; 95% CI: 0.76 to 0.87) [226].
Glucagon-like peptide 1 receptor agonists (GLP-1RAs) have been reported to have both AMPK-activating and mTORC1-inhibitory effects [237, 238]. The LEADER showed that the 3-point MACE occurred in significantly fewer patients in the liraglutide group than in the placebo group (hazard ratio (HR): 0.87; 95% CI: 0.78 to 0.97; P < 0.001) [227]. The SUSTAIN-6 trial showed that the incidence of the 3-point MACE in the semaglutide group (6.6%) was lower than in the placebo group (6.6%) (HR: 0.74; 95% CI: 0.58 to 0.95; P < 0.001) [228]. A meta-analysis of preclinical studies showed that GLP-1RAs improved learning and memory abilities and reduced brain amyloid beta and phosphorylated tau levels in rodents with AD [229]. A meta-analysis of human studies also showed that GLP-1RAs effectively improved cognitive function in AD [230].
SGLT2is and GLP-1RAs effectively lower HbA1c, but the mechanisms by which they do so are quite different, making them a compelling combination. Recently, both antidiabetic drugs have been shown to reduce MACE, although likely through various mechanisms. The combination therapy with SGLT2is and GLP-1RAs on aging-related diseases in patients with type 2 diabetes mellitus may be potentially beneficial [239]. A meta-analysis showed that in real-life conditions, the combination therapy with SGLT2is and GLP-1RAs reduced CVD compared with both GLP-1RA monotherapy and SGLT2i monotherapy [240].
Dual glucose-dependent insulinotropic polypeptide (GIP)/GLP-1RAs have been reported to have both AMPK-activating and mTORC1-inhibitory effects [241, 242]. Further research is needed to understand the impact of GIP/GLP-1RAs on age-related diseases such as CVD and AD.
Mitochondrial dysfunction and oxidative stress are important factors for aging. Imeglimin improves mitochondrial function and reduces reactive oxygen species [243]. Imeglimin has also been reported to enhance AMPK activity [244]; however, the effect of imeglimin on mTORC1 has not yet been reported. Further research is warranted to understand the impact of imeglimin on age-related diseases.
| Future Directions for Application to Aging-Related Diseases | ▴Top |
Cellular senescence is a direct cause of age-related diseases, and senescent cells have been recognized as potential therapeutic targets [245, 246]. Eradication of senescent cells appears to be an excellent way to reduce age-related changes such as inflammation [247]. A promising anti-aging drug cocktail consists of dasatinib (a tyrosine kinase inhibitor) and quercetin (a natural flavonoid) [248]. This cocktail has been examined in animal models of age-related diseases such as atherosclerosis, type 2 diabetes, and AD [247]. The first human study showed a reduction in the number of senescent cells in the adipose tissue of patients with DKD [249]. Recently, the combination of fisetin (a flavonoid) and metformin demonstrated superior anti-aging effects on type 2 diabetes-associated aortic aging compared to metformin monotherapy [250]. Senolytics may be a promising, direct anti-aging therapeutic strategy specifically for diabetes and its complications.
| Limitations of This Review | ▴Top |
The biological links between aging and type 2 diabetes may vary by genetics, ethnicity, lifestyle, and comorbidities, which could not be evaluated in this review. This review does not discuss whether findings are equally applicable across different populations, such as different races. Furthermore, this review could not identify the most susceptible age group for the anti-aging medicine, nor the best time to start anti-aging medicine.
| Conclusions | ▴Top |
Metabolic abnormalities in type 2 diabetes increase factors that accelerate aging, such as inflammation and oxidative stress, and such factors exacerbate metabolic abnormalities and complications of type 2 diabetes. Therefore, it is important to consider the treatment options and mechanisms between aging and type 2 diabetes. A common feature observed in type 2 diabetes and aging is reduced AMPK activity and increased mTORC1 activity, and it is meaningful to focus on this phenomenon in treatments for type 2 diabetes. Exercise has both AMPK-activating and mTORC1-inhibitory effects and has been reported to improve type 2 diabetes and prevent aging-related disease. Nutritional interventions that have both AMPK-activating and mTORC1-inhibitory effects are beneficially associated with type 2 diabetes. Metformin, SGLT2is, and GLP-1RAs, which are anti-diabetic drugs with accumulated evidence of their effectiveness in preventing CV events, also contain both AMPK-activating and mTORC1-inhibitory effects, and are expected to be used in the treatment of AD. Furthermore, senolytics may be a promising, direct anti-aging therapeutic strategy specifically for type 2 diabetes and its complications.
Acknowledgments
We thank Ayano Sakakibara and Yukie Kawamura, the Division of Research Support staff at the National Kohnodai Medical Center, Japan Institute for Health Security.
Financial Disclosure
The authors have no financial disclosures to report.
Conflict of Interest
The authors declare that they have no conflict of interest concerning this article.
Author Contributions
HY contributed to conceptualization, supervision, writing - original draft, review and editing. MH, HK, and HA contributed to formal analysis and data curation. HK contributed to conceptualization and data curation. All authors have read and agreed to the published version of the manuscript.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
AD: Alzheimer’s disease; AGEs: advanced glycation end products; AMPK: adenosine monophosphate-activated protein kinase; BDNF: brain-derived neurotrophic factor; BMD: bone mineral density; CTRP3: C1q/tumor necrosis factor-related protein-3; CI: confidence interval; CVD: cardiovascular diseases; DR: diabetic retinopathy; DKD: diabetic kidney disease; EVs: extracellular vesicles; GIP: glucose-dependent insulinotropic polypeptide; GLP-1RAs: glucagon-like peptide 1 receptor agonists; HFD: high-fat diet; IL-6: interleukin-6; IGF-1: insulin-like growth factor 1; MACE: major adverse cardiovascular events; MD: mean difference; NSN: nutrient-sensing network; mTORC1: mammalian target of rapamycin complex 1; NAD+: nicotinamide adenine dinucleotide; NADPH: nicotinamide adenine dinucleotide phosphate; NAMPT: nicotinamide phosphoribosyl transferase; NF-κB: nuclear factor-kappa B; NLRP3: nod-like receptor pyrin 3; Nrf2: nuclear factor E2-related factor 2; PGC1α: peroxisome proliferator-activated receptor-γ coactivator 1α; PWV: pulse wave velocity; PUFA: polyunsaturated fatty acids; RAGE: receptors for advanced glycation end products; ROS: reactive oxygen species; SASP: senescence-associated secretory phenotype; SGLT2: sodium glucose cotransporter-2; SIRT: sirtuin; TBF: Tang Bi formula; TERT: telomerase reverse transcriptase; ULK1: unc-51-like autophagy activating kinase 1; WAT: white adipose tissue
| References | ▴Top |
- Guo J, Huang X, Dou L, Yan M, Shen T, Tang W, Li J. Aging and
aging-related diseases: from molecular mechanisms to interventions and treatments. Signal
Transduct Target Ther. 2022;7(1):391.
doi pubmed - Li Y, Tian X, Luo J, Bao T, Wang S, Wu X. Molecular mechanisms of
aging and anti-aging strategies. Cell Commun Signal. 2024;22(1):285.
doi pubmed - Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks
of aging: an expanding universe. Cell. 2023;186(2):243-278.
doi pubmed - Kobayashi H, Umetani M, Nishio M, Shigetomi H, Imanaka S, Hashimoto
H. Molecular mechanisms of cellular senescence in age-related endometrial dysfunction. Cells.
2025;14(12):858.
doi pubmed - Schneider JL, Rowe JH, Garcia-de-Alba C, Kim CF, Sharpe AH, Haigis
MC. The aging lung: Physiology, disease, and immunity. Cell. 2021;184(8):1990-2019.
doi pubmed - Zhang Y, Zhang Z, Tu C, Chen X, He R. Advanced glycation end products
in disease development and potential interventions. Antioxidants (Basel).
2025;14(4):492.
doi pubmed - DeJong EN, Surette MG, Bowdish DME. The gut microbiota and unhealthy
aging: disentangling cause from consequence. Cell Host Microbe. 2020;28(2):180-189.
doi pubmed - Alsegiani AS, Shah ZA. The influence of gut microbiota alteration on
age-related neuroinflammation and cognitive decline. Neural Regen Res.
2022;17(11):2407-2412.
doi pubmed - Zhao Y, Simon M, Seluanov A, Gorbunova V. DNA damage and repair in
age-related inflammation. Nat Rev Immunol. 2023;23(2):75-89.
doi pubmed - Stead ER, Bjedov I. Balancing DNA repair to prevent ageing and
cancer. Exp Cell Res. 2021;405(2):112679.
doi pubmed - Huang B, Hu X. Causality of aging hallmarks. Aging Dis. 2025.
doi pubmed - Pang Y, Zhang H, Li H, Wang X, Wei P, Yi L, Lin S. Epigenetic
insights into aging: emerging roles of natural products in therapeutic interventions. Phytother
Res. 2025;39(7):3300-3322.
doi pubmed - Kaltsas A. Multi-omics perspectives on testicular aging: unraveling
germline dysregulation, niche dysfunction, and epigenetic remodeling. Cells.
2025;14(12).
doi pubmed - Holwerda AM, Paulussen KJM, Overkamp M, Goessens JPB, Kramer IF,
Wodzig W, Verdijk LB, et al. Dose-dependent increases in whole-body net protein balance and
dietary protein-derived amino acid incorporation into myofibrillar protein during recovery from
resistance exercise in older men. J Nutr. 2019;149(2):221-230.
doi pubmed - Slack C, Alic N, Foley A, Cabecinha M, Hoddinott MP, Partridge L. The
Ras-Erk-ETS-signaling pathway is a drug target for longevity. Cell. 2015;162(1):72-83.
doi pubmed - Yang BA, Westerhof TM, Sabin K, Merajver SD, Aguilar CA. Engineered
tools to study intercellular communication. Adv Sci (Weinh). 2021;8(3):2002825.
doi pubmed - Fafian-Labora JA, O'Loghlen A. Classical and nonclassical
intercellular communication in senescence and ageing. Trends Cell Biol.
2020;30(8):628-639.
doi pubmed - Zhu D, Li X, Tian Y. Mitochondrial-to-nuclear communication in aging:
an epigenetic perspective. Trends Biochem Sci. 2022;47(8):645-659.
doi pubmed - Zhao Q, Liu J, Deng H, Ma R, Liao JY, Liang H, Hu J, et al. Targeting
mitochondria-located circRNA SCAR alleviates NASH via reducing mROS output. Cell.
2020;183(1):76-93.e22.
doi pubmed - Igarashi M, Miura M, Williams E, Jaksch F, Kadowaki T, Yamauchi T,
Guarente L. NAD(+) supplementation rejuvenates aged gut adult stem cells. Aging Cell.
2019;18(3):e12935.
doi pubmed - Xie N, Zhang L, Gao W, Huang C, Huber PE, Zhou X, Li C, et al. NAD(+)
metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther.
2020;5(1):227.
doi pubmed - Li Q, Qian Z, Huang Y, Yang X, Yang J, Xiao N, Liang G, et al.
Mechanisms of endothelial senescence and vascular aging. Biogerontology. 2025;26(4):128.
doi pubmed - Kalamakis G, Brune D, Ravichandran S, Bolz J, Fan W, Ziebell F,
Stiehl T, et al. Quiescence modulates stem cell maintenance and regenerative capacity in the
aging brain. Cell. 2019;176(6):1407-1419.e1414.
doi pubmed - Lawton A, Tripodi N, Feehan J. Running on empty: Exploring stem cell
exhaustion in geriatric musculoskeletal disease. Maturitas. 2024;188:108066.
doi pubmed - Kaushik S, Tasset I, Arias E, Pampliega O, Wong E, Martinez-Vicente
M, Cuervo AM. Autophagy and the hallmarks of aging. Ageing Res Rev. 2021;72:101468.
doi pubmed - Cassidy LD, Narita M. Autophagy at the intersection of aging,
senescence, and cancer. Mol Oncol. 2022;16(18):3259-3275.
doi pubmed - Aktas G. Exploring the link: Hemogram-derived markers in type 2
diabetes mellitus and its complications. World J Diabetes.
2025;16(7):105233.
doi pubmed - Bilgin S, Aktas G, Kurtkulagi O, Atak BM, Duman TT. Edmonton frail
score is associated with diabetic control in elderly type 2 diabetic subjects. J Diabetes
Metab Disord. 2020;19(1):511-514.
doi pubmed - Akan S, Aktas G. Relationship between frailty, according to three
frail scores, and clinical and laboratory parameters of the geriatric patients with type 2
Diabetes Mellitus. Rev Assoc Med Bras (1992). 2022;68(8):1073-1077.
doi pubmed - Kocak MZ, Aktas G, Atak B, Bilgin S, Kurtkulagi O, Duman TT, Ozcil
IE. The association between vitamin D levels and handgrip strength in elderly men. Acta
Endocrinol (Buchar). 2020;16(2):263-266.
doi pubmed - Salminen A, Kaarniranta K, Kauppinen A. Age-related changes in AMPK
activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling
pathways. Ageing Res Rev. 2016;28:15-26.
doi pubmed - Aydin S, Tekinalp SG, Tuzcu B, Cam F, Sevik MO, Tatar E, Deepak Kalaskarf D, et al. The role of AMP-activated protein kinase activators on energy balance and cellular metabolism in type 2 diabetes mellitus. Obesity Medicine. 2025;53:100577.
- Mannick JB, Lamming DW. Targeting the biology of aging with mTOR
inhibitors. Nat Aging. 2023;3(6):642-660.
doi pubmed - Bar-Tana J. Type 2 diabetes - unmet need, unresolved pathogenesis,
mTORC1-centric paradigm. Rev Endocr Metab Disord. 2020;21(4):613-629.
doi pubmed - Qu Q, Miao Y, Hu Z. Research progress on the regulation of grey hair production by disruption of melanocyte stem cell homeostasis. Chin J Plast Surg. 2017;33:313-316.
- Yang K, Han X. Lipidomics: techniques, applications, and outcomes
related to biomedical sciences. Trends Biochem Sci. 2016;41(11):954-969.
doi pubmed - Wu L, Li S, He C. Lipidomics combined with network pharmacology to
explore differences in the mechanisms of grey hair development between type 2 diabetes mellitus
and normal populations (Female). Int J Mol Sci. 2025;26(5):2034.
doi pubmed - Moraes VR, Melo MO, Maia Campos P. Evaluation of morphological and
structural skin alterations on diabetic subjects by biophysical and imaging techniques. Life
(Basel). 2023;13(2):579.
doi pubmed - Samocha-Bonet D, Wu B, Ryugo DK. Diabetes mellitus and hearing loss:
A review. Ageing Res Rev. 2021;71:101423.
doi pubmed - Antal B, McMahon LP, Sultan SF, Lithen A, Wexler DJ, Dickerson B,
Ratai EM, et al. Type 2 diabetes mellitus accelerates brain aging and cognitive decline:
Complementary findings from UK Biobank and meta-analyses. Elife. 2022;11:e73138.
doi pubmed - Li M, Sun H, Chen H, Ma W, Li Y. Type 2 diabetes and bone mineral
density: A meta-analysis and systematic review. Medicine (Baltimore).
2024;103(45):e40468.
doi pubmed - Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L,
Ducimetiere P, et al. Aortic stiffness is an independent predictor of all-cause and
cardiovascular mortality in hypertensive patients. Hypertension. 2001;37(5):1236-1241.
doi pubmed - Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of
cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and
meta-analysis. J Am Coll Cardiol. 2010;55(13):1318-1327.
doi pubmed - Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM,
Vita JA, et al. Arterial stiffness and cardiovascular events: the Framingham Heart Study.
Circulation. 2010;121(4):505-511.
doi pubmed - Kim HL, Lee JM, Seo JB, Chung WY, Kim SH, Zo JH, Kim MA. The effects
of metabolic syndrome and its components on arterial stiffness in relation to gender.
J Cardiol. 2015;65(3):243-249.
doi pubmed - Masding MG, Stears AJ, Burdge GC, Wootton SA, Sandeman DD.
Premenopausal advantages in postprandial lipid metabolism are lost in women with type 2
diabetes. Diabetes Care. 2003;26(12):3243-3249.
doi pubmed - Madonna R, Balistreri CR, De Rosa S, Muscoli S, Selvaggio S,
Selvaggio G, Ferdinandy P, et al. Impact of sex differences and diabetes on coronary
atherosclerosis and ischemic heart disease. J Clin Med. 2019;8(1):98.
doi pubmed - Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C,
Hayoz D, Pannier B, et al. Expert consensus document on arterial stiffness: methodological
issues and clinical applications. Eur Heart J. 2006;27(21):2588-2605.
doi pubmed - Townsend RR, Wilkinson IB, Schiffrin EL, Avolio AP, Chirinos JA,
Cockcroft JR, Heffernan KS, et al. Recommendations for improving and standardizing vascular
research on arterial stiffness: a scientific statement from the American Heart Association.
Hypertension. 2015;66(3):698-722.
doi pubmed - Yapei Y, Xiaoyan R, Sha Z, Li P, Xiao M, Shuangfeng C, Lexin W, et
al. Clinical significance of arterial stiffness and thickness biomarkers in type 2 diabetes
mellitus: an up-to-date meta-analysis. Med Sci Monit. 2015;21:2467-2475.
doi pubmed - Szablewski L. Changes in cells associated with insulin resistance.
Int J Mol Sci. 2024;25(4):2397.
doi pubmed - Takahashi F, Hashimoto Y, Kaji A, Sakai R, Okamura T, Kitagawa N,
Okada H, et al. Sarcopenia is associated with a risk of mortality in people with type 2 diabetes
mellitus. Front Endocrinol (Lausanne). 2021;12:783363.
doi pubmed - Laohajaroensombat O, Limpaarayakul T, Sathavarodom N, Boonyavarakul
A, Samakkarnthai P. A comparative analysis of sarcopenia screening methods in Thai people with
type 2 diabetes mellitus in an outpatient setting. BMC Geriatr. 2025;25(1):346.
doi pubmed - Iakovou E, Kourti M. A comprehensive overview of the complex role of
oxidative stress in aging, the contributing environmental stressors and emerging antioxidant
therapeutic interventions. Front Aging Neurosci. 2022;14:827900.
doi pubmed - Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction,
and aging. J Signal Transduct. 2012;2012:646354.
doi pubmed - Checa J, Aran JM. Reactive oxygen species: drivers of physiological
and pathological processes. J Inflamm Res. 2020;13:1057-1073.
doi pubmed - Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Base
excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis.
2009;30(1):2-10.
doi pubmed - Erusalimsky JD. Oxidative stress, telomeres and cellular senescence:
What non-drug interventions might break the link? Free Radic Biol Med. 2020;150:87-95.
doi pubmed - Yesupatham A, Saraswathy R. Role of oxidative stress in prediabetes
development. Biochem Biophys Rep. 2025;43:102069.
doi pubmed - Dawi J, Misakyan Y, Affa S, Kades S, Narasimhan A, Hajjar F, Besser
M, et al. Oxidative stress, glutathione insufficiency, and inflammatory pathways in type 2
diabetes mellitus: implications for therapeutic interventions. Biomedicines.
2024;13(1):18.
doi pubmed - Klisic A, Karakasis P, Patoulias D, Khalaji A, Ninic A. Are oxidative
stress biomarkers reliable part of multimarker panel in female patients with type 2 diabetes
mellitus? Metab Syndr Relat Disord. 2024;22(9):679-685.
doi pubmed - Yang Z, Zhang L, Liu J, Li D. Litchi pericarp extract treats type 2
diabetes mellitus by regulating oxidative stress, inflammatory response, and energy metabolism.
Antioxidants (Basel). 2024;13(4):495.
doi pubmed - Singh A, Schurman SH, Bektas A, Kaileh M, Roy R, Wilson DM, 3rd, Sen
R, et al. Aging and inflammation. Cold Spring Harb Perspect Med. 2024;14(6):a041197.
doi pubmed - Pellegrini V, La Grotta R, Carreras F, Giuliani A, Sabbatinelli J,
Olivieri F, Berra CC, et al. Inflammatory trajectory of type 2 diabetes: novel opportunities for
early and late treatment. Cells. 2024;13(19):1662.
doi pubmed - Burhans MS, Hagman DK, Kuzma JN, Schmidt KA, Kratz M. Contribution of
adipose tissue inflammation to the development of type 2 diabetes mellitus. Compr Physiol.
2018;9(1):1-58.
doi pubmed - Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation
as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract.
2014;105(2):141-150.
doi pubmed - Alrouji M, Al-Kuraishy HM, Al-Gareeb AI, Alexiou A, Papadakis M,
Jabir MS, Saad HM, et al. NF-kappaB/NLRP3 inflammasome axis and risk of Parkinson's disease in
Type 2 diabetes mellitus: A narrative review and new perspective. J Cell Mol Med.
2023;27(13):1775-1789.
doi pubmed - Shamsi A, Shahwan M, Husain FM, Khan MS. Characterization of
methylglyoxal induced advanced glycation end products and aggregates of human transferrin:
Biophysical and microscopic insight. Int J Biol Macromol.
2019;138:718-724.
doi pubmed - Li Q, Wen Y, Wang L, Chen B, Chen J, Wang H, Chen L.
Hyperglycemia-induced accumulation of advanced glycosylation end products in fibroblast-like
synoviocytes promotes knee osteoarthritis. Exp Mol Med. 2021;53(11):1735-1747.
doi pubmed - Chaudhuri J, Bains Y, Guha S, Kahn A, Hall D, Bose N, Gugliucci A, et
al. The role of advanced glycation end products in aging and metabolic diseases: bridging
association and causality. Cell Metab. 2018;28(3):337-352.
doi pubmed - Rajaobelina K, Cougnard-Gregoire A, Delcourt C, Gin H,
Barberger-Gateau P, Rigalleau V. Autofluorescence of skin advanced glycation end products:
marker of metabolic memory in elderly population. J Gerontol A Biol Sci Med
Sci. 2015;70(7):841-846.
doi pubmed - Khalid M, Petroianu G, Adem A. Advanced glycation end products and
diabetes mellitus: mechanisms and perspectives. Biomolecules. 2022;12(4).
doi pubmed - Nowotny K, Jung T, Hohn A, Weber D, Grune T. Advanced glycation end
products and oxidative stress in type 2 diabetes mellitus. Biomolecules.
2015;5(1):194-222.
doi pubmed - Kim HJ, Jeong MS, Jang SB. Molecular characteristics of RAGE and
advances in small-molecule inhibitors. Int J Mol Sci. 2021;22(13):6904.
doi pubmed - Mengstie MA, Chekol Abebe E, Behaile Teklemariam A, Tilahun Mulu A,
Agidew MM, Teshome Azezew M, Zewde EA, et al. Endogenous advanced glycation end products in the
pathogenesis of chronic diabetic complications. Front Mol Biosci. 2022;9:1002710.
doi pubmed - Pekar A, Sukumar M. Quantitation of aggregates in therapeutic
proteins using sedimentation velocity analytical ultracentrifugation: practical considerations
that affect precision and accuracy. Anal Biochem. 2007;367(2):225-237.
doi pubmed - Hamjane N, Mechita MB, Nourouti NG, Barakat A. Gut microbiota
dysbiosis -associated obesity and its involvement in cardiovascular diseases and type 2
diabetes. A systematic review. Microvasc Res. 2024;151:104601.
doi pubmed - Yu Y, Ding Y, Wang S, Jiang L. Gut microbiota dysbiosis and its
impact on type 2 diabetes: from pathogenesis to therapeutic strategies. Metabolites.
2025;15(6):397.
doi pubmed - Lazar E, Sherzai A, Adeghate J, Sherzai D. Gut dysbiosis, insulin
resistance and Alzheimer's disease: review of a novel approach to neurodegeneration. Front
Biosci (Schol Ed). 2021;13(1):17-29.
doi pubmed - Smith U, Li Q, Ryden M, Spalding KL. Cellular senescence and its role
in white adipose tissue. Int J Obes (Lond). 2021;45(5):934-943.
doi pubmed - Franzke B, Schwingshackl L, Wagner KH. Chromosomal damage measured by
the cytokinesis block micronucleus cytome assay in diabetes and obesity - A systematic review
and meta-analysis. Mutat Res Rev Mutat Res. 2020;786:108343.
doi pubmed - Chakravarti D, LaBella KA, DePinho RA. Telomeres: history, health,
and hallmarks of aging. Cell. 2021;184(2):306-322.
doi pubmed - Victorelli S, Passos JF. Telomeres and cell senescence - size matters
not. EBioMedicine. 2017;21:14-20.
doi pubmed - Shammas MA. Telomeres, lifestyle, cancer, and aging. Curr Opin Clin
Nutr Metab Care. 2011;14(1):28-34.
doi pubmed - Tamura Y, Takubo K, Aida J, Araki A, Ito H. Telomere attrition and
diabetes mellitus. Geriatr Gerontol Int. 2016;16(Suppl 1):66-74.
doi pubmed - Sampson MJ, Hughes DA. Chromosomal telomere attrition as a mechanism
for the increased risk of epithelial cancers and senescent phenotypes in type 2 diabetes.
Diabetologia. 2006;49(8):1726-1731.
doi pubmed - Bure IV, Nemtsova MV, Kuznetsova EB. Histone modifications and
non-coding RNAs: mutual epigenetic regulation and role in pathogenesis. Int J Mol
Sci. 2022;23(10):5801.
doi pubmed - Sherazi SAM, Abbasi A, Jamil A, Uzair M, Ikram A, Qamar S, Olamide
AA, et al. Molecular hallmarks of long non-coding RNAs in aging and its significant effect on
aging-associated diseases. Neural Regen Res. 2023;18(5):959-968.
doi pubmed - Yi SJ, Kim K. New insights into the role of histone changes in aging.
Int J Mol Sci. 2020;21(21):8241.
doi pubmed - Miller JL, Grant PA. The role of DNA methylation and histone
modifications in transcriptional regulation in humans. Subcell Biochem. 2013;61:289-317.
doi pubmed - Gharipour M, Mani A, Amini Baghbahadorani M, de Souza Cardoso CK,
Jahanfar S, Sarrafzadegan N, de Oliveira C, et al. How are epigenetic modifications related to
cardiovascular disease in older adults? Int J Mol Sci. 2021;22(18).
doi pubmed - Long Y, Mao C, Liu S, Tao Y, Xiao D. Epigenetic modifications in
obesity-associated diseases. MedComm (2020). 2024;5(2):e496.
doi pubmed - Ronn T, Ofori JK, Perfilyev A, Hamilton A, Pircs K, Eichelmann F,
Garcia-Calzon S, et al. Genes with epigenetic alterations in human pancreatic islets impact
mitochondrial function, insulin secretion, and type 2 diabetes. Nat Commun.
2023;14(1):8040.
doi pubmed - Odimegwu CL, Uwaezuoke SN, Chikani UN, Mbanefo NR, Adiele KD, Nwolisa
CE, Eneh CI, et al. Targeting the epigenetic marks in type 2 diabetes mellitus: will epigenetic
therapy be a valuable adjunct to pharmacotherapy? Diabetes Metab Syndr Obes.
2024;17:3557-3576.
doi pubmed - Ahmed I, Chakraborty R, Faizy AF, Moin S. Exploring the key role of
DNA methylation as an epigenetic modulator in oxidative stress related islet cell injury in
patients with type 2 diabetes mellitus: a review. J Diabetes Metab Disord.
2024;23(2):1699-1718.
doi pubmed - Vasishta S, Ammankallu S, Poojary G, Gomes SM, Ganesh K, Umakanth S,
Adiga P, et al. High glucose induces DNA methyltransferase 1 dependent epigenetic reprogramming
of the endothelial exosome proteome in type 2 diabetes. Int J Biochem Cell Biol.
2024;176:106664.
doi pubmed - Li DL, Hodge AM, Southey MC, Giles GG, Dugue PA. Association of
epigenetic markers of aging with prevalent and incident type 2 diabetes.
J Gerontol A Biol Sci Med Sci. 2025;80(7):glaf085.
doi pubmed - Fry CS, Rasmussen BB. Skeletal muscle protein balance and metabolism
in the elderly. Curr Aging Sci. 2011;4(3):260-268.
doi pubmed - Wall BT, Gorissen SH, Pennings B, Koopman R, Groen BB, Verdijk LB,
van Loon LJ. Aging Is Accompanied by a Blunted Muscle Protein Synthetic Response to Protein
Ingestion. PLoS One. 2015;10(11):e0140903.
doi pubmed - Katsanos CS, Kobayashi H, Sheffield-Moore M, Aarsland A, Wolfe RR.
Aging is associated with diminished accretion of muscle proteins after the ingestion of a small
bolus of essential amino acids. Am J Clin Nutr. 2005;82(5):1065-1073.
doi pubmed - Cuthbertson D, Smith K, Babraj J, Leese G, Waddell T, Atherton P,
Wackerhage H, et al. Anabolic signaling deficits underlie amino acid resistance of wasting,
aging muscle. FASEB J. 2005;19(3):422-424.
doi pubmed - Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and
chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem.
2009;78:959-991.
doi pubmed - Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular
mechanisms and promising therapies. Nat Rev Drug Discov. 2015;14(1):58-74.
doi pubmed - Andersen H, Nielsen S, Mogensen CE, Jakobsen J. Muscle strength in
type 2 diabetes. Diabetes. 2004;53(6):1543-1548.
doi pubmed - Park SW, Goodpaster BH, Strotmeyer ES, Kuller LH, Broudeau R,
Kammerer C, de Rekeneire N, et al. Accelerated loss of skeletal muscle strength in older adults
with type 2 diabetes: the health, aging, and body composition study. Diabetes Care.
2007;30(6):1507-1512.
doi pubmed - Yang K, Hou R, Zhao J, Wang X, Wei J, Pan X, Zhu X. Lifestyle effects
on aging and CVD: A spotlight on the nutrient-sensing network. Ageing Res Rev.
2023;92:102121.
doi pubmed - Green CL, Lamming DW, Fontana L. Molecular mechanisms of dietary
restriction promoting health and longevity. Nat Rev Mol Cell Biol. 2022;23(1):56-73.
doi pubmed - Yang G, Cao X, Li X, Zhang J, Ma C, Zhang N, Lu Q, et al. Association
of unhealthy lifestyle and childhood adversity with acceleration of aging among UK biobank
participants. JAMA Netw Open. 2022;5(9):e2230690.
doi pubmed - Ribeiro-Rodrigues TM, Kelly G, Korolchuk VI, Girao H. Intercellular communication and aging. Aging Fundam Biol Soc. 2023;2023:257-274.
- Akbar N, Azzimato V, Choudhury RP, Aouadi M. Extracellular vesicles
in metabolic disease. Diabetologia. 2019;62(12):2179-2187.
doi pubmed - Rutter GA, Georgiadou E, Martinez-Sanchez A, Pullen TJ. Metabolic and
functional specialisations of the pancreatic beta cell: gene disallowance, mitochondrial
metabolism and intercellular connectivity. Diabetologia. 2020;63(10):1990-1998.
doi pubmed - Guo Y, Guan T, Shafiq K, Yu Q, Jiao X, Na D, Li M, et al.
Mitochondrial dysfunction in aging. Ageing Res Rev. 2023;88:101955.
doi pubmed - Pinti MV, Fink GK, Hathaway QA, Durr AJ, Kunovac A, Hollander JM.
Mitochondrial dysfunction in type 2 diabetes mellitus: an organ-based analysis.
Am J Physiol Endocrinol Metab. 2019;316(2):E268-E285.
doi pubmed - Iheagwam FN, Joseph AJ, Adedoyin ED, Iheagwam OT, Ejoh SA.
Mitochondrial dysfunction in diabetes: shedding light on a widespread oversight.
Pathophysiology. 2025;32(1):9.
doi pubmed - Prolla TA, Denu JM. NAD+ deficiency in age-related mitochondrial
dysfunction. Cell Metab. 2014;19(2):178-180.
doi pubmed - McReynolds MR, Chellappa K, Baur JA. Age-related NAD(+) decline. Exp
Gerontol. 2020;134:110888.
doi pubmed - Covarrubias AJ, Perrone R, Grozio A, Verdin E. NAD(+) metabolism and
its roles in cellular processes during ageing. Nat Rev Mol Cell Biol.
2021;22(2):119-141.
doi pubmed - Manickam R, Santhana S, Xuan W, Bisht K, Tipparaju S. Nampt: a new
therapeutic target for modulating NAD(+) levels in metabolic, cardiovascular, and
neurodegenerative diseases. Can J Physiol Pharmacol. 2025;103(7):208-224.
doi pubmed - Ruskovska T, Bernlohr DA. The role of NAD(+) in metabolic regulation
of adipose tissue: implications for obesity-induced insulin resistance. Biomedicines.
2023;11(9).
doi pubmed - Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular
senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol.
2021;22(2):75-95.
doi pubmed - Zhang L, Pitcher LE, Yousefzadeh MJ, Niedernhofer LJ, Robbins PD, Zhu
Y. Cellular senescence: a key therapeutic target in aging and diseases. J Clin Invest.
2022;132(15):e158450.
doi pubmed - Aguayo-Mazzucato C, Andle J, Lee TB, Jr., Midha A, Talemal L,
Chipashvili V, Hollister-Lock J, et al. Acceleration of beta Cell Aging Determines Diabetes and
Senolysis Improves Disease Outcomes. Cell Metab. 2019;30(1):129-142 e124.
doi pubmed - Yan X, Gao Z, Zhou Y, Gao F, Li Q. Expressions of IGF-1R and Ki-67 in
breast cancer patients with diabetes mellitus and an analysis of biological characteristics.
Pak J Med Sci. 2022;38(1):281-286.
doi pubmed - Tuduri E, Soriano S, Almagro L, Garcia-Heredia A, Rafacho A,
Alonso-Magdalena P, Nadal A, et al. The effects of aging on male mouse pancreatic beta-cell
function involve multiple events in the regulation of secretion: influence of insulin
sensitivity. J Gerontol A Biol Sci Med Sci. 2022;77(3):405-415.
doi pubmed - Krstic J, Reinisch I, Schupp M, Schulz TJ, Prokesch A. p53 functions
in adipose tissue metabolism and homeostasis. Int J Mol Sci.
2018;19(9):2622.
doi pubmed - Murakami T, Inagaki N, Kondoh H. Cellular senescence in diabetes
mellitus: distinct senotherapeutic strategies for adipose tissue and pancreatic beta cells.
Front Endocrinol (Lausanne). 2022;13:869414.
doi pubmed - Mi L, Hu J, Li N, Gao J, Huo R, Peng X, Zhang N, et al. The mechanism
of stem cell aging. Stem Cell Rev Rep. 2022;18(4):1281-1293.
doi pubmed - Terenzi DC, Trac JZ, Teoh H, Gerstein HC, Bhatt DL, Al-Omran M, Verma
S, et al. Vascular regenerative cell exhaustion in diabetes: translational opportunities to
mitigate cardiometabolic risk. Trends Mol Med. 2019;25(7):640-655.
doi pubmed - Fadini GP, Ciciliot S, Albiero M. Concise review: perspectives and
clinical implications of bone marrow and circulating stem cell defects in diabetes. Stem Cells.
2017;35(1):106-116.
doi pubmed - Aman Y, Schmauck-Medina T, Hansen M, Morimoto RI, Simon AK, Bjedov I,
Palikaras K, et al. Autophagy in healthy aging and disease. Nat Aging.
2021;1(8):634-650.
doi pubmed - Metaxakis A, Ploumi C, Tavernarakis N. Autophagy in age-associated
neurodegeneration. Cells. 2018;7(5):37.
doi pubmed - Jung HS, Lee MS. Macroautophagy in homeostasis of pancreatic
beta-cell. Autophagy. 2009;5(2):241-243.
doi pubmed - Ding Y, Choi ME. Autophagy in diabetic nephropathy.
J Endocrinol. 2015;224(1):R15-30.
doi pubmed - Vivot K, Pasquier A, Goginashvili A, Ricci R. Breaking bad and
breaking good: beta-cell autophagy pathways in diabetes. J Mol Biol.
2020;432(5):1494-1513.
doi pubmed - Muralidharan C, Conteh AM, Marasco MR, Crowder JJ, Kuipers J, de Boer
P, Linnemann AK. Pancreatic beta cell autophagy is impaired in type 1 diabetes. Diabetologia.
2021;64(4):865-877.
doi pubmed - Salemkour Y, Lenoir O. Endothelial autophagy dysregulation in
diabetes. Cells. 2023;12(6):947.
doi pubmed - Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, et al.
Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial
biogenesis. Cell Metab. 2007;5(2):151-156.
doi pubmed - Sorrenti V, Benedetti F, Buriani A, Fortinguerra S, Caudullo G,
Davinelli S, Zella D, et al. Immunomodulatory and antiaging mechanisms of resveratrol,
rapamycin, and metformin: focus on mTOR and AMPK signaling networks. Pharmaceuticals (Basel).
2022;15(8):912.
doi pubmed - Ramamurthy S, Ronnett GV. Developing a head for energy sensing:
AMP-activated protein kinase as a multifunctional metabolic sensor in the brain.
J Physiol. 2006;574(Pt 1):85-93.
doi pubmed - Heidrich F, Schotola H, Popov AF, Sohns C, Schuenemann J, Friedrich
M, Coskun KO, et al. AMPK - activated protein kinase and its role in energy metabolism of the
heart. Curr Cardiol Rev. 2010;6(4):337-342.
doi pubmed - Bodine SC. The role of mTORC1 in the regulation of skeletal muscle
mass. Fac Rev. 2022;11:32.
doi pubmed - Deblon N, Bourgoin L, Veyrat-Durebex C, Peyrou M, Vinciguerra M,
Caillon A, Maeder C, et al. Chronic mTOR inhibition by rapamycin induces muscle insulin
resistance despite weight loss in rats. Br J Pharmacol.
2012;165(7):2325-2340.
doi pubmed - Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin. Annu Rev
Immunol. 1996;14:483-510.
doi pubmed - de la Cruz Lopez KG, Toledo Guzman ME, Sanchez EO, Garcia Carranca A.
mTORC1 as a regulator of mitochondrial functions and a therapeutic target in cancer. Front
Oncol. 2019;9:1373.
doi pubmed - Han X, Goh KY, Lee WX, Choy SM, Tang HW. The importance of
mTORC1-autophagy axis for skeletal muscle diseases. Int J Mol Sci.
2022;24(1):297.
doi pubmed - Scepanovic G, Balaghi N, Rothenberg KE, Fernandez-Gonzalez R. mTor
limits autophagy to facilitate cell volume expansion and rapid wound repair in Drosophila
embryos. Dev Cell. 2025;60(10):1400-1410.e1403.
doi pubmed - Yin X, Guo Z, Song C. AMPK, a key molecule regulating aging-related
myocardial ischemia-reperfusion injury. Mol Biol Rep. 2024;51(1):257.
doi pubmed - Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic
disease. Cell Metab. 2011;13(1):11-22.
doi pubmed - Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK)
controls the aging process via an integrated signaling network. Ageing Res Rev.
2012;11(2):230-241.
doi pubmed - Hosseini Y, Niknejad A, Sabbagh Kashani A, Gholami M, Roustaie M,
Mohammadi M, Momtaz S, et al. NLRP3 inflammasomes pathway: a key target for Metformin.
Inflammopharmacology. 2025;33(4):1729-1760.
doi pubmed - Fernandez E, Aragones G, Renneburg C, Francisco SG, Kageyama S, Komatsu M, Taylor A. Targeting mTOR pathway to ameliorate glycation-derived proteotoxicity in retinal and lens cells. Invest Ophthalmol Vis Sci. 2020;61:3099.
- Sun X, Zhu MJ. AMP-activated protein kinase: a therapeutic target in
intestinal diseases. Open Biol. 2017;7(8):170104.
doi pubmed - Gongol B, Sari I, Bryant T, Rosete G, Marin T. AMPK: an epigenetic
landscape modulator. Int J Mol Sci. 2018;19(10):3238.
doi pubmed - Ding N, Maiuri AR, O'Hagan HM. The emerging role of epigenetic
modifiers in repair of DNA damage associated with chronic inflammatory diseases. Mutat Res Rev
Mutat Res. 2019;780:69-81.
doi pubmed - Smith EM, Pendlebury DF, Nandakumar J. Structural biology of
telomeres and telomerase. Cell Mol Life Sci. 2020;77(1):61-79.
doi pubmed - Sung JY, Kim SG, Park SY, Kim JR, Choi HC. Telomere stabilization by
metformin mitigates the progression of atherosclerosis via the AMPK-dependent p-PGC-1alpha
pathway. Exp Mol Med. 2024;56(9):1967-1979.
doi pubmed - Wang C, Wang H, Zhang D, Luo W, Liu R, Xu D, Diao L, et al.
Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to
macroautophagy. Nat Commun. 2018;9(1):3492.
doi pubmed - Dossou AS, Basu A. The emerging roles of mTORC1 in macromanaging
autophagy. Cancers (Basel). 2019;11(10):1422.
doi pubmed - Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial
homeostasis. Nat Rev Mol Cell Biol. 2018;19(2):121-135.
doi pubmed - Li W, Sauve AA. NAD(+) content and its role in mitochondria. Methods
Mol Biol. 2015;1241:39-48.
doi pubmed - Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC,
Elliott PJ, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1
activity. Nature. 2009;458(7241):1056-1060.
doi pubmed - Begum MK, Konja D, Singh S, Chlopicki S, Wang Y. Endothelial SIRT1 as
a target for the prevention of arterial aging: promises and challenges. J Cardiovasc
Pharmacol. 2021;78(Suppl 6):S63-S77.
doi pubmed - Qi W, Hu C, Zhao D, Li X. SIRT1-SIRT7 in diabetic kidney disease:
biological functions and molecular mechanisms. Front Endocrinol (Lausanne).
2022;13:801303.
doi pubmed - Sun C, Bai S, Liang Y, Liu D, Liao J, Chen Y, Zhao X, et al. The role
of Sirtuin 1 and its activators in age-related lung disease. Biomed Pharmacother.
2023;162:114573.
doi pubmed - He S, Wang Y, Liu J, Li P, Luo X, Zhang B. Activating SIRT1
deacetylates NF-kappaB p65 to alleviate liver inflammation and fibrosis via inhibiting NLRP3
pathway in macrophages. Int J Med Sci. 2023;20(4):505-519.
doi pubmed - Peng Y, Yao SY, Chen Q, Jin H, Du MQ, Xue YH, Liu S. True or false?
Alzheimer's disease is type 3 diabetes: Evidences from bench to bedside. Ageing Res Rev.
2024;99:102383.
doi pubmed - Yerra VG, Areti A, Kumar A. Adenosine monophosphate-activated protein
kinase abates hyperglycaemia-induced neuronal injury in experimental models of diabetic
neuropathy: effects on mitochondrial biogenesis, autophagy and neuroinflammation. Mol Neurobiol.
2017;54(3):2301-2312.
doi pubmed - Roy Chowdhury SK, Smith DR, Saleh A, Schapansky J, Marquez A, Gomes
S, Akude E, et al. Impaired adenosine monophosphate-activated protein kinase signalling in
dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in
diabetes. Brain. 2012;135(Pt 6):1751-1766.
doi pubmed - Yang C, Yu H, Zhou L, Su H, Li X, Qi W, Lian F. Tang Bi formula
alleviates diabetic sciatic neuropathy via AMPK/PGC-1alpha/MFN2 pathway activation. Sci Rep.
2025;15(1):25069.
doi pubmed - Zhang CH, Lv X, Du W, Cheng MJ, Liu YP, Zhu L, Hao J. The Akt/mTOR
cascade mediates high glucose-induced reductions in BDNF via DNMT1 in Schwann cells in diabetic
peripheral neuropathy. Exp Cell Res. 2019;383(1):111502.
doi pubmed - Noori T, Sureda A, Shirooie S. Role of natural mTOR inhibitors in
treatment of diabetes mellitus. Fundam Clin Pharmacol. 2023;37(3):461-479.
doi pubmed - Yan Z, Wang C, Meng Z, Gan L, Guo R, Liu J, Bond Lau W, et al.
C1q/TNF-related protein 3 prevents diabetic retinopathy via AMPK-dependent stabilization of
blood-retinal barrier tight junctions. Cells. 2022;11(5):779.
doi pubmed - Lee SHS, Lee JY, Choi JS, Kim HJ, Kim J, Cha S, Lee KJ, et al. mTOR
inhibition as a novel gene therapeutic strategy for diabetic retinopathy. PLoS One.
2022;17(6):e0269951.
doi pubmed - Han YC, Tang SQ, Liu YT, Li AM, Zhan M, Yang M, Song N, et al. AMPK
agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and
streptozotocin induced diabetic mice. Cell Death Dis. 2021;12(10):925.
doi pubmed - Shi J, Liu X, Jiao Y, Tian J, An J, Zou G, Zhuo L. mTOR pathway: A
key player in diabetic nephropathy progression and therapeutic targets. Genes Dis.
2025;12(2):101260.
doi pubmed - Yasuda-Yamahara M, Kume S, Maegawa H. Roles of mTOR in diabetic
kidney disease. Antioxidants (Basel). 2021;10(2):321.
doi pubmed - Heidary Moghaddam R, Samimi Z, Asgary S, Mohammadi P, Hozeifi S,
Hoseinzadeh-Chahkandak F, Xu S, et al. Natural AMPK activators in cardiovascular disease
prevention. Front Pharmacol. 2021;12:738420.
doi pubmed - Kaldirim M, Lang A, Pfeiler S, Fiegenbaum P, Kelm M, Bonner F, Gerdes
N. Modulation of mTOR signaling in cardiovascular disease to target acute and chronic
inflammation. Front Cardiovasc Med. 2022;9:907348.
doi pubmed - Guo J, Wu Y, Wan Z, Zhang Z. Post-translational modifications
orchestrate mTOR-driven cell death in cardiovascular disease. Front Cardiovasc Med.
2025;12:1620669.
doi pubmed - Wang L, Li N, Shi FX, Xu WQ, Cao Y, Lei Y, Wang JZ, et al.
Upregulation of AMPK ameliorates Alzheimer's disease-like tau pathology and memory impairment.
Mol Neurobiol. 2020;57(8):3349-3361.
doi pubmed - Mueed Z, Tandon P, Maurya SK, Deval R, Kamal MA, Poddar NK. Tau and
mTOR: the hotspots for multifarious diseases in Alzheimer's development. Front Neurosci.
2018;12:1017.
doi pubmed - Rapaka D, Bitra VR, Challa SR, Adiukwu PC. mTOR signaling as a
molecular target for the alleviation of Alzheimer's disease pathogenesis. Neurochem Int.
2022;155:105311.
doi pubmed - Pataky MW, Young WF, Nair KS. Hormonal and metabolic changes of aging
and the influence of lifestyle modifications. Mayo Clin Proc. 2021;96(3):788-814.
doi pubmed - Dennison EM. Osteoarthritis: The importance of hormonal status in
midlife women. Maturitas. 2022;165:8-11.
doi pubmed - Yang X, Yan K, Zhang Q, Yusufu A, Ran J. Meta-analysis of estrogen in
osteoarthritis: clinical status and protective effects. Altern Ther Health Med.
2023;29(1):224-230.
pubmed - Coryell PR, Diekman BO, Loeser RF. Mechanisms and therapeutic
implications of cellular senescence in osteoarthritis. Nat Rev Rheumatol.
2021;17(1):47-57.
doi pubmed - Ahmad Hairi H, Ibrahim NI, Sadikan MZ, Jayusman PA, Shuid AN.
Deciphering the role of classical oestrogen receptor in insulin resistance and type 2 diabetes
mellitus: From molecular mechanism to clinical evidence. Bioimpacts. 2025;15:30378.
doi pubmed - Anagnostis P, Christou K, Artzouchaltzi AM, Gkekas NK, Kosmidou N,
Siolos P, Paschou SA, et al. Early menopause and premature ovarian insufficiency are associated
with increased risk of type 2 diabetes: a systematic review and meta-analysis.
Eur J Endocrinol. 2019;180(1):41-50.
doi pubmed - Dorman JS, Steenkiste AR, Foley TP, Strotmeyer ES, Burke JP, Kuller
LH, Kwoh CK, et al. Menopause in type 1 diabetic women: is it premature? Diabetes.
2001;50(8):1857-1862.
doi pubmed - Thong EP, Codner E, Laven JSE, Teede H. Diabetes: a metabolic and
reproductive disorder in women. Lancet Diabetes Endocrinol. 2020;8(2):134-149.
doi pubmed - Zhu A, Andino J, Daignault-Newton S, Chopra Z, Sarma A, Dupree JM.
What is a normal testosterone level for young men? Rethinking the 300 ng/dL cutoff for
testosterone deficiency in men 20-44 years old. J Urol. 2022;208(6):1295-1302.
doi pubmed - Wittert G, Grossmann M. Obesity, type 2 diabetes, and testosterone in
ageing men. Rev Endocr Metab Disord. 2022;23(6):1233-1242.
doi pubmed - Hasan H, Bhushan S, Fijak M, Meinhardt A. Mechanism of inflammatory
associated impairment of sperm function, spermatogenesis and steroidogenesis. Front Endocrinol
(Lausanne). 2022;13:897029.
doi pubmed - Khalil SHA, Dandona P, Osman NA, Assaad RS, Zaitoon BTA, Almas AA,
Amin NG. Diabetes surpasses obesity as a risk factor for low serum testosterone level. Diabetol
Metab Syndr. 2024;16(1):143.
doi pubmed - Ramachandran P, Zitzmann M, König CS, Mulhern J, Ramachandran S, Hackett G. Testosterone undecanoate is associated with improved ageing male symptoms score in men with type 2 diabetes and adult-onset testosterone deficiency: re-analyzed results from a randomised controlled trial. Explor Endocr Metab Dis. 2024;1:177-190.
- Richter EA, Ruderman NB. AMPK and the biochemistry of exercise:
implications for human health and disease. Biochem J. 2009;418(2):261-275.
doi pubmed - Watson K, Baar K. mTOR and the health benefits of exercise. Semin
Cell Dev Biol. 2014;36:130-139.
doi pubmed - Ma JC, Shu S, Chen TX, Bai HJ, Yang Y, Ding XW. Intervention effect
of combined resistance and aerobic exercise on type 2 diabetes: A meta-analysis.
World J Diabetes. 2025;16(7):108121.
doi pubmed - Shokri K, Karimian A, Radfar A, Mohammadi A, Amerizadeh A, Karimi R,
Sadeghi M. Effect of exercise-based cardiac rehabilitation in patients with acute coronary
syndrome: a systematic review and meta-analysis. BMC Sports Sci Med Rehabil.
2025;17(1):233.
doi pubmed - Gai Y, Dai X, Qian M, Lin G, Pan P, Dai T, Luo Y, et al. Effects of
physical exercise on cognitive and motor function in patients with Alzheimer's disease: a
meta-analysis based on randomized controlled trials. Cogn Neurodyn. 2025;19(1):133.
doi pubmed - Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K,
Laboy R, et al. Dietary restriction and AMPK increase lifespan via mitochondrial network and
peroxisome remodeling. Cell Metab. 2017;26(6):884-896.e885.
doi pubmed - Tulsian R, Velingkaar N, Kondratov R. Caloric restriction effects on
liver mTOR signaling are time-of-day dependent. Aging (Albany NY). 2018;10(7):1640-1648.
doi pubmed - Jayedi A, Zeraattalab-Motlagh S, Shahinfar H, Gregg EW, Shab-Bidar S.
Effect of calorie restriction in comparison to usual diet or usual care on remission of type 2
diabetes: a systematic review and meta-analysis of randomized controlled trials.
Am J Clin Nutr. 2023;117(5):870-882.
doi pubmed - Pan J, Li Z, Zhu M, Guo L, Chen W, Yu L. Vitamin E exerts a
mitigating effect on LPS-induced acute lung injury by regulating macrophage polarization through
the AMPK/NRF2/NF-kappaB pathway. Int Immunopharmacol. 2025;159:114893.
doi pubmed - Yu Y, Hou L, Song H, Xu P, Sun Y, Wu K. Akt/AMPK/mTOR pathway was
involved in the autophagy induced by vitamin E succinate in human gastric cancer SGC-7901 cells.
Mol Cell Biochem. 2017;424(1-2):173-183.
doi pubmed - Asbaghi O, Nazarian B, Yousefi M, Anjom-Shoae J, Rasekhi H, Sadeghi
O. Effect of vitamin E intake on glycemic control and insulin resistance in diabetic patients:
an updated systematic review and meta-analysis of randomized controlled trials. Nutr J.
2023;22(1):10.
doi pubmed - Huang J, Min S, Hong R, Zou M, Zhou D. High-dose Vitamin C inhibits
PD-L1 expression by activating AMPK in colorectal cancer. Immunobiology.
2025;230(3):152893.
doi pubmed - Qin S, Wang G, Chen L, Geng H, Zheng Y, Xia C, Wu S, et al.
Pharmacological vitamin C inhibits mTOR signaling and tumor growth by degrading Rictor and
inducing HMOX1 expression. PLoS Genet. 2023;19(2):e1010629.
doi pubmed - Nosratabadi S, Ashtary-Larky D, Hosseini F, Namkhah Z, Mohammadi S,
Salamat S, Nadery M, et al. The effects of vitamin C supplementation on glycemic control in
patients with type 2 diabetes: A systematic review and meta-analysis. Diabetes Metab Syndr.
2023;17(8):102824.
doi pubmed - Lan F, Weikel KA, Cacicedo JM, Ido Y. Resveratrol-induced
AMP-activated protein kinase activation is cell-type dependent: lessons from basic research for
clinical application. Nutrients. 2017;9(7):751.
doi pubmed - Park D, Jeong H, Lee MN, Koh A, Kwon O, Yang YR, Noh J, et al.
Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci Rep.
2016;6:21772.
doi pubmed - Abdelhaleem IA, Brakat AM, Adayel HM, Asla MM, Rizk MA, Aboalfetoh
AY. The effects of resveratrol on glycemic control and cardiometabolic parameters in patients
with T2DM: A systematic review and meta-analysis. Med Clin (Barc). 2022;158(12):576-585.
doi pubmed - Lu S, Zhang Y, Yu W. Activation of the AMPK-mTOR pathway by
astaxanthin against cold ischemia-reperfusion in rat liver. Tohoku J Exp Med.
2025;265(3):151-159.
doi pubmed - Urakaze M, Kobashi C, Satou Y, Shigeta K, Toshima M, Takagi M,
Takahashi J, et al. The beneficial effects of astaxanthin on glucose metabolism and modified
low-density lipoprotein in healthy volunteers and subjects with prediabetes. Nutrients.
2021;13(12):4381.
doi pubmed - Oppedisano F, Macri R, Gliozzi M, Musolino V, Carresi C, Maiuolo J,
Bosco F, et al. The anti-inflammatory and antioxidant properties of n-3 PUFAs: their role in
cardiovascular protection. Biomedicines. 2020;8(9):306.
doi pubmed - Nie J, Chen J, Yang J, Pei Q, Li J, Liu J, Xu L, et al. Inhibition of
mammalian target of rapamycin complex 1 signaling by n-3 polyunsaturated fatty acids promotes
locomotor recovery after spinal cord injury. Mol Med Rep. 2018;17(4):5894-5902.
doi pubmed - Qian F, Ardisson Korat AV, Imamura F, Marklund M, Tintle N, Virtanen
JK, Zhou X, et al. n-3 fatty acid biomarkers and incident type 2 diabetes: an individual
participant-level pooling project of 20 prospective cohort studies. Diabetes Care.
2021;44(5):1133-1142.
doi pubmed - Miller SL, Wolfe RR. The danger of weight loss in the elderly.
J Nutr Health Aging. 2008;12(7):487-491.
doi pubmed - Bonora BM, Avogaro A, Fadini GP. Euglycemic ketoacidosis. Curr Diab
Rep. 2020;20(7):25.
doi pubmed - Wang L, Huang X, Sun M, Zheng T, Zheng L, Lin X, Ruan J, et al. New
light on omega-3 polyunsaturated fatty acids and diabetes debate: a population
pharmacokinetic-pharmacodynamic modelling and intake threshold study. Nutr Diabetes.
2024;14(1):8.
doi pubmed - Bahardoust M, Mousavi S, Yariali M, Haghmoradi M, Hadaegh F, Khalili
D, Delpisheh A. Effect of metformin (vs. placebo or sulfonylurea) on all-cause and
cardiovascular mortality and incident cardiovascular events in patients with diabetes: an
umbrella review of systematic reviews with meta-analysis. J Diabetes Metab Disord.
2024;23(1):27-38.
doi pubmed - Luo A, Ning P, Lu H, Huang H, Shen Q, Zhang D, Xu F, et al.
Association between metformin and Alzheimer's disease: a systematic review and meta-analysis of
clinical observational studies. J Alzheimers Dis. 2022;88(4):1311-1323.
doi pubmed - Petrie JR. Metformin beyond type 2 diabetes: emerging and potential
new indications. Diabetes Obes Metab. 2024;26(Suppl 3):31-41.
doi pubmed - Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S,
Mattheus M, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes.
N Engl J Med. 2015;373(22):2117-2128.
doi pubmed - Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N,
Shaw W, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes.
N Engl J Med. 2017;377(7):644-657.
doi pubmed - Kim HK, Biessels GJ, Yu MH, Hong N, Lee YH, Lee BW, Kang ES, et al.
SGLT2 inhibitor use and risk of dementia and Parkinson disease among patients with type 2
diabetes. Neurology. 2024;103(8):e209805.
doi pubmed - Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck
MA, Nissen SE, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes.
N Engl J Med. 2016;375(4):311-322.
doi pubmed - Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jodar E, Leiter LA,
Lingvay I, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes.
N Engl J Med. 2016;375(19):1834-1844.
doi pubmed - Kong F, Wu T, Dai J, Zhai Z, Cai J, Zhu Z, Xu Y, et al. Glucagon-like
peptide 1 (GLP-1) receptor agonists in experimental Alzheimer's disease models: a systematic
review and meta-analysis of preclinical studies. Front Pharmacol. 2023;14:1205207.
doi pubmed - Bi Z, Wang L, Wang W. Evaluating the effects of glucagon-like
peptide-1 receptor agonists on cognitive function in Alzheimer's disease: A systematic review
and meta-analysis. Adv Clin Exp Med. 2023;32(11):1223-1231.
doi pubmed - Goel S, Singh R, Singh V, Singh H, Kumari P, Chopra H, Sharma R, et
al. Metformin: Activation of 5' AMP-activated protein kinase and its emerging potential beyond
anti-hyperglycemic action. Front Genet. 2022;13:1022739.
doi pubmed - Amin S, Lux A, O'Callaghan F. The journey of metformin from glycaemic
control to mTOR inhibition and the suppression of tumour growth. Br J Clin
Pharmacol. 2019;85(1):37-46.
doi pubmed - Chalmers J, Cooper ME. UKPDS and the legacy effect.
N Engl J Med. 2008;359(15):1618-1620.
doi pubmed - Hasanvand A. The role of AMPK-dependent pathways in cellular and
molecular mechanisms of metformin: a new perspective for treatment and prevention of diseases.
Inflammopharmacology. 2022;30(3):775-788.
doi pubmed - Safaie N, Masoumi S, Alizadeh S, Mirzajanzadeh P, Nejabati HR,
Hajiabbasi M, Alivirdiloo V, et al. SGLT2 inhibitors and AMPK: The road to cellular
housekeeping? Cell Biochem Funct. 2024;42(1):e3922.
doi pubmed - Schaub JA, AlAkwaa FM, McCown PJ, Naik AS, Nair V, Eddy S, Menon R,
et al. SGLT2 inhibitors mitigate kidney tubular metabolic and mTORC1 perturbations in
youth-onset type 2 diabetes. J Clin Invest. 2023;133(5):e164486.
doi pubmed - Zhang Y, Chen H, Feng Y, Liu M, Lu Z, Hu B, Chen L, et al. Activation
of AMPK by GLP-1R agonists mitigates Alzheimer-related phenotypes in transgenic mice. Nat Aging.
2025;5(6):1097-1113.
doi pubmed - Parichatikanond W, Pandey S, Mangmool S. Exendin-4 exhibits
cardioprotective effects against high glucose-induced mitochondrial abnormalities: Potential
role of GLP-1 receptor and mTOR signaling. Biochem Pharmacol. 2024;229:116552.
doi pubmed - DeFronzo RA. Combination therapy with GLP-1 receptor agonist and
SGLT2 inhibitor. Diabetes Obes Metab. 2017;19(10):1353-1362.
doi pubmed - Scheen AJ. Cardiovascular and renal effects of the combination
therapy of a GLP-1 receptor agonist and an SGLT2 inhibitor in observational real-life studies.
Diabetes Metab. 2025;51(1):101594.
doi pubmed - Xu J, Chen P, Wu D, Zhou Q, Chen S, Ding X, Xiong H. The novel
GLP-1/GIP dual agonist DA3-CH improves rat type 2 diabetes through activating AMPK/ACC signaling
pathway. Aging (Albany NY). 2023;15(20):11152-11161.
doi pubmed - Chen M, Zhao N, Shi W, Xing Y, Liu S, Meng X, Li L, et al.
Glucose-dependent insulinotropic polypeptide/glucagon-like peptide 1 receptor agonist
tirzepatide promotes branched chain amino acid catabolism to prevent myocardial infarction in
non-diabetic mice. Cardiovasc Res. 2025;121(3):454-467.
doi pubmed - Yanai H, Adachi H, Hakoshima M, Katsuyama H. Glucose-lowering effects
of imeglimin and its possible beneficial effects on diabetic complications. Biology (Basel).
2023;12(5):726.
doi pubmed - Hozumi K, Sugawara K, Ishihara T, Ishihara N, Ogawa W. Effects of
imeglimin on mitochondrial function, AMPK activity, and gene expression in hepatocytes. Sci Rep.
2023;13(1):746.
doi pubmed - Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness
RA, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature.
2016;530(7589):184-189.
doi pubmed - Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de
Sluis B, Kirkland JL, et al. Clearance of p16Ink4a-positive senescent cells delays
ageing-associated disorders. Nature. 2011;479(7372):232-236.
doi pubmed - Sikora E, Bielak-Zmijewska A, Mosieniak G. Targeting normal and
cancer senescent cells as a strategy of senotherapy. Ageing Res Rev. 2019;55:100941.
doi pubmed - Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N,
Palmer AK, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs.
Aging Cell. 2015;14(4):644-658.
doi pubmed - Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N,
Hashmi SK, Herrmann SM, et al. Senolytics decrease senescent cells in humans: Preliminary report
from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease.
EBioMedicine. 2019;47:446-456.
doi pubmed - Ji XM, Dong XX, Li JP, Tai GJ, Qiu S, Wei W, Silumbwe CW, et al.
Fisetin clears senescent cells through the Pi3k-Akt-Bcl-2/Bcl-xl pathway to alleviate diabetic
aortic aging. Phytother Res. 2025;39(6):2757-2775.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Clinical Medicine Research is published by Elmer Press Inc.