| Journal of Clinical Medicine Research, ISSN 1918-3003 print, 1918-3011 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Med Res and Elmer Press Inc |
| Journal website https://jocmr.elmerjournals.com |
Case Report
Volume 17, Number 12, December 2025, pages 733-739

Bilateral Adrenalectomy for Primary Bilateral Macronodular Adrenocortical Hyperplasia With a Novel ARMC5 Mutation Site
Cai Guo Yua, e, Nan Nan Wub, c, e, Bin Caob, c, Hao Lin Gongb, c, Yan Mab, c, Shao Zhong Xiand, Bao Yu Zhanga, Jing Keb, c, f, Dong Zhaob, c, f
aDepartment of Endocrinology, Beijing Anzhen Hospital, Capital Medical University, Beijing 101118, China
bCenter for Endocrine Metabolism and Immune Diseases, Beijing Luhe Hospital, Capital Medical University, Beijing 101149, China
cBeijing Key Laboratory of Diabetes Research and Care, Beijing 100000, China
dDepartment of Urology, Beijing Luhe Hospital, Capital Medical University, Beijing 101149, China
eThese authors contributed equally to the article.
fCorresponding Authors: Jing Ke and Dong Zhao, Center for Endocrine Metabolism and Immune Diseases, Beijing Luhe Hospital, Capital Medical University, Beijing 101149, Chinaand
Manuscript submitted August 31, 2025, accepted November 26, 2025, published online December 24, 2025
Short title: PBMAH Patient With a Novel ARMC5 Mutation Site
doi: https://doi.org/10.14740/jocmr6359
| Abstract | ▴Top |
Primary bilateral macronodular adrenocortical hyperplasia (PBMAH) is a heterogeneous disorder, with clinical spectrum ranging from subclinical to severe overt Cushing syndrome (CS). Armadillo repeat containing 5 (ARMC5) mutations, found in 20-55% of cases, are often associated with more severe disease, and specific genotypes may correlate with distinct phenotypes. A 60-year-old woman was admitted with a 1-year history of progressive weight gain and hypokalemia, alongside a 13-year history of refractory hypertension. She exhibited classic CS features and metabolic complications, including hypertension and diabetes. Laboratory tests revealed elevated cortisol, suppressed adrenocorticotropic hormone (ACTH), and positive low- and high-dose dexamethasone suppression tests. Contrast-enhanced computed tomography (CT) showed bilateral irregular macronodular adrenal masses. Adrenal venous sampling (AVS) demonstrated no lateralization (lateralization index < 4). Whole-exome sequencing of peripheral blood leukocytes identified a novel ARMC5 mutation (c.534_555dup, p.Ser186Profs*19). A right adrenalectomy was performed in 2023. Although symptoms improved, cortisol levels remained significantly elevated. Consequently, a left adrenalectomy was performed 15 months later in 2024, which led to marked improvement in her CS symptoms, biomarker levels, and metabolic complications. This case report describes a novel ARMC5 mutation site in a PBMAH patient who ultimately required bilateral adrenalectomy. While AVS combined with CT can help determine the dominant side for surgery, patients with ARMC5 mutations and symmetrically sized tumors often require bilateral adrenalectomy for cure.
Keywords: Primary bilateral macronodular adrenocortical hyperplasia; ARMC5; Adrenal venous sampling; Cushing syndrome
| Introduction | ▴Top |
Primary bilateral macronodular adrenocortical hyperplasia (PBMAH) is a clinically heterogeneous disorder characterized by bilateral adrenal macronodules > 1.0 cm with variable cortisol secretion, ranging from asymptomatic subclinical hypercortisolism to overt Cushing syndrome (CS) [1]. It is also named bilateral macronodular adrenocortical disease according to the updated World Health Organization (WHO) Classification of Endocrine and Neuroendocrine Tumors (2025). Overt cases present with classic features including weight gain, proximal myopathy, reddish striae, facial plethora, buffalo hump, skin fragility, easy bruising, and hyperandrogenism, along with metabolic complications (hypertension, diabetes, venous thromboembolism) and skeletal manifestations (osteopenia, osteoporosis) [2]. Most symptomatic patients are diagnosed between 40 and 60 years of age, often after years of subtle progression [1], while milder cases are frequently detected incidentally during abdominal imaging for unrelated conditions [2].
The bilateral nature and occasional familial occurrence suggest a genetic basis [2], with germline mutations in tumor suppressor genes (MEN1, APC, FH) causing syndromic forms associated with various neoplasms [2, 3]. Armadillo repeat containing 5 (ARMC5) mutations occur in 20-55% of PBMAH cases [1, 4, 5] and correlate with more severe phenotypes including earlier onset, larger nodules (> 4 cm), higher cortisol levels, and greater need for surgical intervention [2, 4]. Emerging evidence suggests that certain clinical features may be mutation-specific [6].
The management of patients with PBMAH has consistently posed clinical challenges, particularly regarding the choice between unilateral and bilateral adrenalectomy, as well as the prevention and management of postoperative adrenal crisis. For patients with significant asymmetry in bilateral nodule size, recent guidelines recommend prioritizing the resection of the larger adrenal gland to reduce the risk of postoperative adrenal crisis. However, for patients with bilaterally similar nodule sizes, it remains uncertain whether unilateral adrenalectomy can achieve effective cure.
Therefore, we report a novel germline ARMC5 mutation site in a PBMAH patient, who ultimately required bilateral adrenalectomy for cure. This case highlights the importance of genetic testing, appropriate surgical plans and continuous follow-up.
| Case Report | ▴Top |
Clinical features
A 60-year-old woman was admitted to our hospital with progressive weight gain and hypokalemia for 1 year, weight increased from 67 kg (body mass index (BMI): 25.8 kg/m2) to 77 kg (BMI: 29.7 kg/m2), blood potassium fluctuated between 3.05 and 3.79 mmol/L, coexisting with refractory hypertension for 13 years (150 - 180/80 - 90 mm Hg), taking four kinds of oral antihypertensive drugs including diuretics for 1 year. In addition, she had obviously high blood glucose levels, with fasting blood glucose levels higher than 10 mmol/L and postprandial glucose levels higher than 17.4 mmol/L, and with poor response to anti-diabetic treatment including continuous subcutaneous insulin pump. Physical examination revealed characteristic CS features, including moon facies, centripetal obesity, abdominal subcutaneous hemorrhages and peripheral edema with cutaneous ecchymoses. Biochemical evaluation demonstrated markedly elevated serum cortisol levels and significantly increased urinary free cortisol (UFC) excretion (Table 1). In addition, her plasma adrenocorticotropic hormone (ACTH) levels were markedly suppressed at all time points (< 1.5 pg/mL). The results of low-dosed dexamethasone suppression test (LD-DST) and high-dosed dexamethasone suppression test (HD-DST) were both positive (Table 2). ACTH-independent hypercortisolism was suggested by the above results. Contrast-enhanced computed tomography (CT) showed bilateral adrenal multiple irregular macronodular masses (right adrenal: 5.7 × 5.0 × 5.6 cm, left adrenal: 5.8 × 4.9 × 6.4 cm) (Fig. 1). Positron emission tomography/computed tomography (PET/CT) revealed multiple nodules in the bilateral adrenal masses, with mutual fusion and uneven increase in fluorodeoxyglucose (FDG) metabolism. Adrenal venous sampling (AVS) was conducted to evaluate the predominant side of cortisol production (Table 3). Selectivity indexes (SI) were 4.15 and 8.38 in the left and right adrenals, respectively, indicating the success of AVS cannulation (SI cutoff of 2.0) [7]. However, there was no predominant side according to the lateralization index (LI = 2.65, SI cutoff of 4) (Table 2). According to approximately equivalent sizes on both sides, we conducted a multidisciplinary discussion with experts from the Department of Urology and the Department of Radiology. Based on the patient’s CT imaging and vascular observations during AVS, it was suggested that the right adrenal gland might have more abundant blood supply. In consideration of the patient’s preferences (she strongly demanded unilateral surgery), the urology team proceeded with the right adrenalectomy first. Pathological examinations and immunohistochemical staining revealed adrenal nodular hyperplasia (Fig. 2). One week after surgery, her serum and UFC levels reduced significantly (Table 1). During the follow-up, the CS symptoms improved, especially the blood pressure and glucose levels, with only nifedipine and dapagliflozin for treatments. Blood potassium levels increased to normal. Symptoms including subcutaneous ecchymosis, limb edema and insomnia disappeared. However, the serum and urinary cortisol levels remained consistently higher than normal during the 6-month and 1-year postoperative follow-up, with ACTH still suppressed lower than normal (Table 1). After our repeated recommendations, the patient ultimately decided to undergo a second surgery and underwent left adrenal subtotal resection.
 Click to view | Table 1. The Main Laboratory and Hormone Expression Profile |
Click to view | Table 2. The Results of LD-DST and HD-DST |
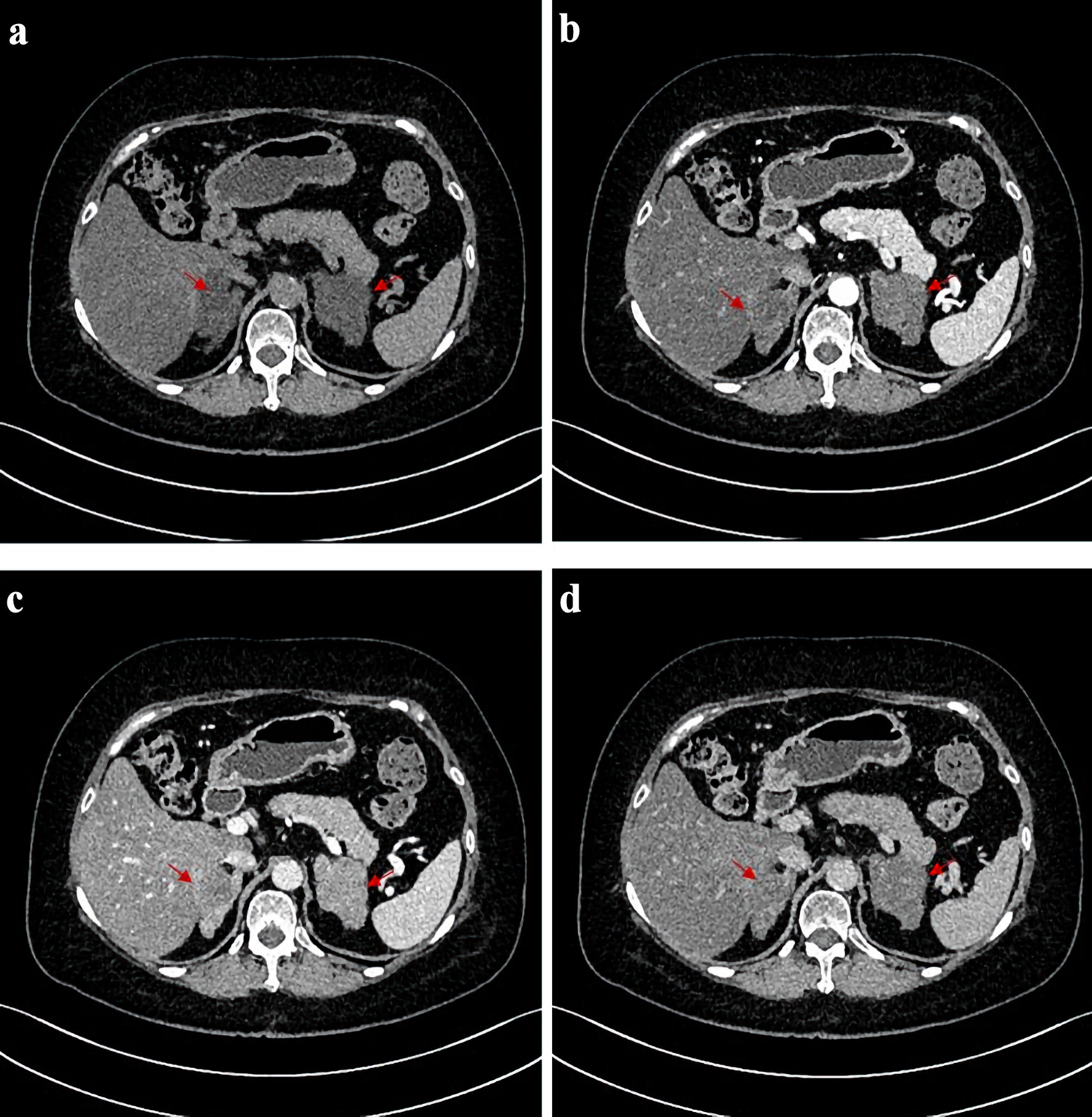 Click for large image | Figure 1. CT scans before operation. CT images of the patient before adrenalectomy. (a) Image of plain CT scan. (b) Image of arterial phase of contrast-enhanced CT. (c) Image of venous phase of contrast-enhanced CT. (d) Image of delayed phase of contrast-enhanced CT. The red arrows point to the position corresponding to bilateral adrenal nodules on both sides. CT: computed tomography. |
Click to view | Table 3. Biochemical Results of AVS |
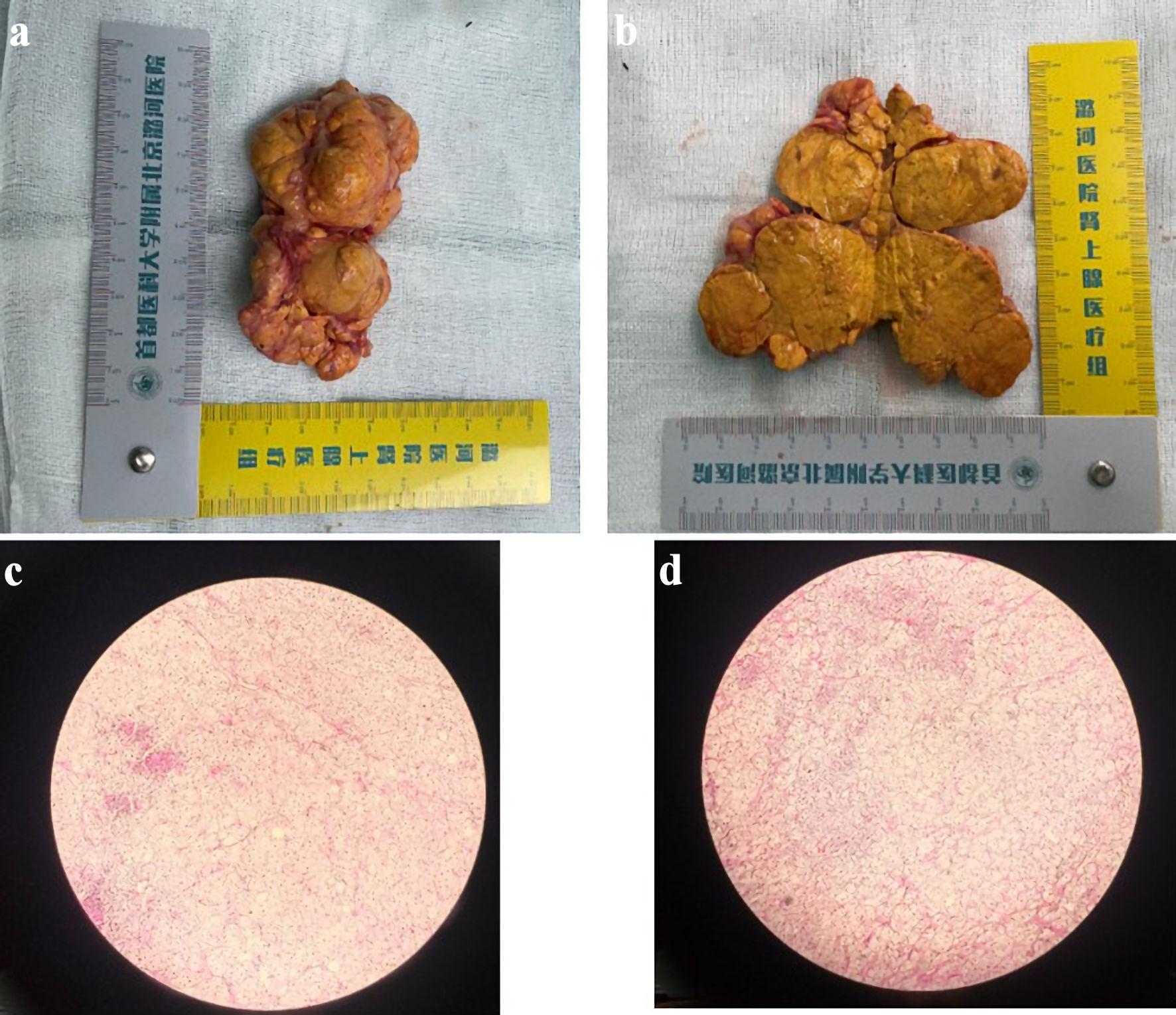 Click for large image | Figure 2. Representative gross and microscopic images of the adrenalectomy specimen. (a, b) Gross images showing a large adrenal nodule with appearance resembling a piece of ginger. (c, d) Corresponding microscopic images (× 10). |
Identification of the mutations
Whole exome sequencing was conducted on peripheral blood leukocytes. A novel heterozygous germline mutation in ARMC5 (c.534_555dup, p.Ser186Profs*19) was identified in peripheral blood leukocytes. This mutation will result in a frameshift mutation in the amino acid encoding the protein from position 186 (serine mutation to proline), and translation is terminated after a frameshift mutation of 18 amino acids. None of her family members had any history of endocrine disease, and all of them refused to have their genes tested.
Ethics approval
All procedures were carried out in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This study was reviewed and approved by the Ethics Committee in the Beijing Luhe Hospital.
| Discussion | ▴Top |
PBMAH is an autosomal dominant disorder marked by the presence of multiple bilateral adrenal nodules, often described as having “a bunch of grapes appearance [7].” In this case, we describe a PBMAH patient exhibiting pronounced CS manifestations, such as facial rounding, truncal obesity, skin bruising, peripheral swelling, and common metabolic disturbances including elevated blood pressure and impaired glucose metabolism. Sizes of bilateral tumors are similar, both larger than 5 cm. No dominant side was found by AVS. A novel pathogenic mutation site in ARMC5 was confirmed by whole exome sequencing (c.534_555dup, p.Ser186Profs*19). Right side of total followed by left subtotal adrenalectomy was conducted for this patient.
In our patient, PBMAH was diagnosed according to four key criteria [8]: 1) Clinical features of CS, such as truncal obesity, high blood pressure, violaceous striae, and hyperglycemia; 2) Biochemical confirmation of hypercortisolism, evidenced by heightened plasma cortisol, elevated UFC, loss of diurnal cortisol variation, and low ACTH; 3) Imaging findings of bilateral adrenal macronodules (> 1.0 cm) on CT; 4) Histopathological evidence of nodular adrenal hyperplasia. The markedly reduced ACTH levels aided in differentiating PBMAH from ACTH-dependent etiologies (e.g., Cushing disease or ectopic ACTH production) [9, 10]. Bilateral macronodules and obviously larger adrenal glands help distinguish PBMAH from other ACTH-independent disorders presenting with CS and bilateral adrenal involvement, such as primary pigmented nodular adrenal disease (PPNAD).
PBMAH displays marked clinical variability, presenting with a spectrum of cortisol secretion ranging from subclinical to overt CS. A significant proportion of PBMAH cases go undiagnosed until overt CS develops, while subclinical forms often escape detection unless adrenal enlargement is incidentally noted during imaging studies [11]. This diversity reflects the condition’s wide phenotypic spectrum. Mounting evidence indicates clear genotype-phenotype associations in PBMAH [11], with ARMC5 mutation carriers typically showing earlier disease onset, higher rates of overt CS, and increased hypertension prevalence [11]. This report of a patient with an ARMC5 mutation who presented with severe CS and metabolic complications (hypertension and diabetes), corroborates the link between this genotype and aggressive phenotypes. Current studies demonstrate that adrenal mass size correlates with both cortisol levels and CS severity [12, 13]. Notably, considerable adrenal enlargement appears necessary for the development of hypercortisolism, which explains why overt CS primarily occurs in PBMAH patients with large adrenal masses [14]. Furthermore, the intensity of cortisol hypersecretion in ARMC5-mutated PBMAH patients correlates with age at clinical presentation [15].
The ARMC5 gene, located at chromosome 16p11.2, comprises six exons and has over 80 documented mutations without established hot spots [2]. These mutations are detected in up to 55% of surgically treated PBMAH patients [11, 16]. Our case revealed a novel heterozygous germline ARMC5 mutation (c.534_555dup, p.Ser186Profs*19) in exon 3 of transcript NM_001288767.1. This frameshift mutation alters the protein sequence from position 186 (serine to proline substitution) and introduces premature termination after 18 aberrant amino acids, potentially leading to truncated protein production or nonsense-mediated mRNA decay. Notably, the Human Gene Mutation Database (HGMD) documents multiple loss-of-function variants downstream of this mutation site associated with clinical manifestations including diabetes mellitus. According to Knudson’s “two-hit” hypothesis for tumor suppressors [1], complete ARMC5 inactivation would require a secondary somatic mutation complementing the germline defect. Supporting this, previous studies have identified nodule-specific somatic ARMC5 mutations in adrenal tissues [1, 17], suggesting their potential role in macronodule formation, though the precise mechanisms underlying mutation-mediated PBMAH pathogenesis remain to be fully elucidated.
The management of PBMAH depends on cortisol secretion status. Patients without biochemical evidence of hypercortisolism at diagnosis should undergo active surveillance with annual clinical and biochemical evaluations. Conversely, patients manifesting overt CS or clinically significant hypercortisolism-related complications (e.g., hypertension, diabetes mellitus, osteoporosis) require consideration of therapeutic intervention. Medical management options include steroidogenesis inhibitors (metyrapone, ketoconazole [18]), glucocorticoid receptor antagonists (mifepristone [19]), and adrenolytic therapy (mitotane [20]). While bilateral total adrenalectomy has traditionally been the definitive surgical treatment for PBMAH with CS [21], it necessitates lifelong glucocorticoid and mineralocorticoid replacement and carries inherent risks of adrenal insufficiency crises. Unilateral adrenalectomy offers reduced perioperative risks [22] but may be complicated by recurrent hypercortisolism [14, 23]. Emerging evidence supports adrenal-sparing approaches involving total resection of the larger gland with contralateral subtotal adrenalectomy as a potentially superior option [24]; however, surgical decision-making remains challenging regarding both selection of the initial side for resection and determination of optimal residual tissue volume. AVS could help localize predominant cortisol secretion sides and guide the decision of adrenalectomy. However, AVS may not be able to effectively distinguish the dominant secretory side for PBMAH patients with ARMC5 mutations and bilateral nodules of similar sizes, like the patient in our case.
Among the more than 10 reported cases with ARMC5 mutations, management has involved either unilateral or bilateral adrenalectomy. A summary of the clinical features from these representative cases is provided here (Supplementary Material 1, jocmr.elmerjournals.com).
Conclusions
In conclusion, a novel ARMC5 mutation site was found in our patients, who presented with similarly sized bilateral adrenal macronodules, severe CS symptoms, and serious metabolic complications. Although the patient initially strongly preferred unilateral surgery, long-term clinical follow-up indicated that bilateral surgery was necessary to achieve definitive cure. This case indicates that AVS combined with CT image could be helpful to determine the dominant side for adrenalectomy; however, patients with ARMC5 mutation and similar tumor sizes usually require bilateral adrenalectomy for cure.
| Supplementary Material | ▴Top |
Suppl 1. Recent cases of primary bilateral macronodular adrenal hyperplasia (2020 - 2025).
Acknowledgments
We thank the patient for her cooperation in the preparation of this case report.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Informed Consent
Written informed consent for publication of identifying images or other personal or clinical details was obtained from the patient.
Author Contributions
Study design was conducted by JK and DZ. Analysis of data was performed by CGY and NNW. AVS was performed by BC and HLG, and adrenalectomy by SZX. Patient management was conducted by CGY, JK, NNW, BYZ and YM. CGY wrote the manuscript which was revised and approved by all authors.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
ARMC5: armadillo repeat containing 5; PBMAH: primary bilateral macronodular adrenal hyperplasia; CS: Cushing syndrome; CT: computed tomography; AVS: adrenal venous sampling; LD-DST: low-dosed dexamethasone suppression test; HD-DST: high-dosed dexamethasone suppression test
| References | ▴Top |
- Assie G, Libe R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, Barreau O, et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing's syndrome. N Engl J Med. 2013;369(22):2105-2114.
doi pubmed - Chevalier B, Vantyghem MC, Espiard S. Bilateral adrenal hyperplasia: pathogenesis and treatment. Biomedicines. 2021;9(10):1397.
doi pubmed - Bouys L, Chiodini I, Arlt W, Reincke M, Bertherat J. Update on primary bilateral macronodular adrenal hyperplasia (PBMAH). Endocrine. 2021;71(3):595-603.
doi pubmed - Faucz FR, Zilbermint M, Lodish MB, Szarek E, Trivellin G, Sinaii N, Berthon A, et al. Macronodular adrenal hyperplasia due to mutations in an armadillo repeat containing 5 (ARMC5) gene: a clinical and genetic investigation. J Clin Endocrinol Metab. 2014;99(6):E1113-1119.
doi pubmed - Bouys L, Vaczlavik A, Jouinot A, Vaduva P, Espiard S, Assie G, Libe R, et al. Identification of predictive criteria for pathogenic variants of primary bilateral macronodular adrenal hyperplasia (PBMAH) gene ARMC5 in 352 unselected patients. Eur J Endocrinol. 2022;187(1):123-134.
doi pubmed - Albiger NM, Regazzo D, Rubin B, Ferrara AM, Rizzati S, Taschin E, Ceccato F, et al. A multicenter experience on the prevalence of ARMC5 mutations in patients with primary bilateral macronodular adrenal hyperplasia: from genetic characterization to clinical phenotype. Endocrine. 2017;55(3):959-968.
doi pubmed - Verma A, Mohan S, Gupta A. ACTH-independent macronodular adrenal hyperplasia: imaging findings of a rare condition : A case report. Abdom Imaging. 2008;33(2):225-229.
doi pubmed - Zhang F, Lin X, Yu X. Primary macronodular adrenal hyperplasia (PMAH) can be generated by a new ARMC5 germline variant (c.52C>T (p.Gln18X)). Endocr J. 2020;67(12):1179-1186.
doi pubmed - Sohaib SA, Hanson JA, Newell-Price JD, Trainer PJ, Monson JP, Grossman AB, Besser GM, et al. CT appearance of the adrenal glands in adrenocorticotrophic hormone-dependent Cushing's syndrome. AJR Am J Roentgenol. 1999;172(4):997-1002.
doi pubmed - Pivonello R, De Martino MC, De Leo M, Lombardi G, Colao A. Cushing's syndrome. Endocrinol Metab Clin North Am. 2008;37(1):135-149, ix.
doi pubmed - Espiard S, Drougat L, Libe R, Assie G, Perlemoine K, Guignat L, Barrande G, et al. ARMC5 mutations in a large cohort of primary macronodular adrenal hyperplasia: clinical and functional consequences. J Clin Endocrinol Metab. 2015;100(6):E926-935.
doi pubmed - Drougat L, Espiard S, Bertherat J. Genetics of primary bilateral macronodular adrenal hyperplasia: a model for early diagnosis of Cushing's syndrome? Eur J Endocrinol. 2015;173(4):M121-131.
doi pubmed - Rubinstein G, Osswald A, Braun LT, Vogel F, Kroiss M, Pilz S, Deniz S, et al. The role of adrenal venous sampling (AVS) in primary bilateral macronodular adrenocortical hyperplasia (PBMAH): a study of 16 patients. Endocrine. 2022;76(2):434-445.
doi pubmed - Debillon E, Velayoudom-Cephise FL, Salenave S, Caron P, Chaffanjon P, Wagner T, Massoutier M, et al. Unilateral adrenalectomy as a first-line treatment of Cushing's syndrome in patients with primary bilateral macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 2015;100(12):4417-4424.
doi pubmed - Kyo C, Usui T, Kosugi R, Torii M, Yonemoto T, Ogawa T, Kotani M, et al. ARMC5 alterations in primary macronodular adrenal hyperplasia (PMAH) and the clinical state of variant carriers. J Endocr Soc. 2019;3(10):1837-1846.
doi pubmed - Vassiliadi DA, Tsagarakis S. Diagnosis and management of primary bilateral macronodular adrenal hyperplasia. Endocr Relat Cancer. 2019;26(10):R567-R581.
doi pubmed - Correa R, Zilbermint M, Berthon A, Espiard S, Batsis M, Papadakis GZ, Xekouki P, et al. The ARMC5 gene shows extensive genetic variance in primary macronodular adrenocortical hyperplasia. Eur J Endocrinol. 2015;173(4):435-440.
doi pubmed - Debono M, Harrison RF, Chadarevian R, Gueroult C, Abitbol JL, Newell-Price J. Resetting the abnormal circadian cortisol rhythm in adrenal incidentaloma patients with mild autonomous cortisol secretion. J Clin Endocrinol Metab. 2017;102(9):3461-3469.
doi pubmed - Cohan P, East HE, Galati SJ, Mercado JU, Lim PJ, Lamerson M, Smith JJ, et al. Mifepristone treatment in four cases of primary bilateral macronodular adrenal hyperplasia (BMAH). J Clin Endocrinol Metab. 2019;104(12):6279-6290.
doi pubmed - Nagai M, Narita I, Omori K, Komura S, Arakawa M. Adrenocorticotropic hormone-independent bilateral adrenocortical macronodular hyperplasia treated with mitotane. Intern Med. 1999;38(12):969-973.
doi pubmed - Guerin C, Taieb D, Treglia G, Brue T, Lacroix A, Sebag F, Castinetti F. Bilateral adrenalectomy in the 21st century: when to use it for hypercortisolism? Endocr Relat Cancer. 2016;23(2):R131-142.
doi pubmed - Powell AC, Stratakis CA, Patronas NJ, Steinberg SM, Batista D, Alexander HR, Pingpank JF, et al. Operative management of Cushing syndrome secondary to micronodular adrenal hyperplasia. Surgery. 2008;143(6):750-758.
doi pubmed - Osswald A, Quinkler M, Di Dalmazi G, Deutschbein T, Rubinstein G, Ritzel K, Zopp S, et al. Long-term outcome of primary bilateral macronodular adrenocortical hyperplasia after unilateral adrenalectomy. J Clin Endocrinol Metab. 2019;104(7):2985-2993.
doi pubmed - Yoshiaki Tanno F, Srougi V, Almeida MQ, Ide Yamauchi F, Morbeck Almeida Coelho F, Nishi MY, Claudia Nogueira Zerbini M, et al. A new insight into the surgical treatment of primary macronodular adrenal hyperplasia. J Endocr Soc. 2020;4(8):bvaa083.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Medicine Research is published by Elmer Press Inc.